
Challenges in Conducting Studies in Chronic Graft-versus-Host Disease
Peer review is under the responsibility of IACH
- DOI
- 10.2991/chi.d.190314.001How to use a DOI?
- Keywords
- GvHD; GRFS; FFS; Response criteria; Composite endpoints
- Abstract
The lack of standardized criteria for measuring therapeutic response has been a major obstacle to the development of therapeutic trials in chronic graft-versus-host disease (cGvHD). Nevertheless, recent advances have been made in understanding of the biology and pathophysiology of cGvHD, as well as establishing more precise criteria for the diagnosis and classification of disease manifestations. The momentum has shifted, and currently there is a long list of new potential treatment targets being identified for cGvHD. Consequently, new drugs are being implemented for its prophylaxis and treatment. It is crucial to continue that trend and develop better systems to test new drugs in clinical practice that would eventually translate toward seeking regulatory review and approval. We provide a historical perspective and current challenges in conducting cGvHD clinical trials.
- Copyright
- © 2019 International Academy for Clinical Hematology. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
Allogeneic hematopoietic cell transplantation (alloHCT) is a potentially curative therapy for people with aggressive or refractory hematologic malignancies. Over 26,000 alloHCT are performed annually worldwide [1]. Chronic graft-versus-host disease (cGvHD) is an immune-mediated, multisystem disorder characterized by immunosuppression and immune dysregulation, increased risk of infections, reduced functionality, and significant impairment in health-related quality of life (HRQOL) in people who are otherwise cured from their cancer [2, 3]. cGvHD is the leading cause of long-term nonrelapse morbidity and mortality after alloHCT, occurring in approximately 40–60% of long-term survivors [4–6]. Steroids are the most commonly used first-line therapy for cGvHD, in spite of their multiple toxicities. Treatments for steroid-refractory cGvHD are still profoundly unsatisfactory. The development of well-designed clinical trials to explore more effective therapies for cGvHD is an urgent, unmet, clinical need.
2. HISTORICAL PERSPECTIVE
Historically, cGvHD was defined by the Seattle group in the early 1980s, before cyclosporine was in use, as any GvHD presenting beyond day 100 after alloHCT [7]. cGvHD was further classified as limited (localized skin involvement and/or liver dysfunction) or extensive (generalized skin involvement, liver histology showing aggressive hepatitis, or involvement of any other target organ). This classification provided little information on the type and severity of the organs involved, was poorly reproducible among investigators, and had limited value in predicting late transplant-related mortality. Subsequently, several other prognostic scales have been developed for predicting the latter; however, none of them provides enough information about the extent of the disease, organs involved, or functional impairment [8–12].
In 2005, the National Institutes of Health (NIH) Consensus Conference consolidated expert opinions and standardized recommendations for the diagnosis and staging, histopathology, biomarkers, response criteria, ancillary therapy and supportive care, and design of clinical trials [13–18]. The cGvHD Consortium in the United States and the German–Austrian–Swiss Consortium in Europe were subsequently established to conduct multicenter studies in cGvHD, and many retrospective and prospective longitudinal studies have been conducted, further validating the NIH criteria [19–25]. In 2014, experts met again, with 9 years of experience, to update the NIH criteria, improve on the original recommendations, clarify controversies, and provide greater specificity and more accurate measures of the disease burden [26–31]. The 2014 NIH Consensus Conference included representatives from academia, regulatory agencies, industry, professional societies, and advocacy groups, and the 2014 NIH criteria represent the most comprehensive, detailed, and widely accepted criteria for cGvHD currently available. However, many of the terms still lack clear and uniform definitions. There are continuous efforts by the NIH, Center for International Blood and Marrow Transplant Research, and European Society for Blood and Marrow Transplantation (EBMT) to review the existing guidelines, address ambiguities, and develop more reliable terminology [32–37].
3. GOALS OF TREATMENT FOR cGvHD
Although cGvHD is known to be the main contributor to late posttransplant morbidity and mortality, it is associated with an important graft-versus-tumor (GVT) effect in patients with hematologic malignancies. Overly aggressive immunosuppression may attenuate GVT reaction and increase the risk of recurrence. How to separate the beneficial GVT effect from harmful cGvHD, and use them optimally to the patient's advantage, is one of major challenges in alloHCT research. The goal of treatment for cGvHD is to stop destructive immunological processes, control the disease activity, and establish immunological tolerance. Consequently, treatment should reduce symptom burden, prevent further organ damage, and improve overall survival and HRQOL. Manifestations of cGvHD wax and wane when immunosuppression is reduced, and the goal is to calibrate treatment to the minimum needed to control the disease. Ultimately, approximately 50% of people will be able to achieve immune tolerance of the graft and discontinue systemic immunosuppression within 7 years posttransplant, 10% will need systemic immunosuppression beyond 7 years (likely indefinitely), and 40% will die from recurrent malignancy or nonrelapse causes [38]. Even though novel cGvHD treatment strategies usually initiate from single-center studies, such trials are frequently limited to a small number of patients, a highly selected patient population, and particular center practices [39]. Larger, prospective, multicenter studies are ultimately needed to identify and verify baseline risk factors associated with outcomes and to test new agents.
4. CHALLENGES IN DIAGNOSING cGvHD
4.1. Applying the NIH Guidelines to Clinical Practice
The NIH guidelines were originally developed for research purposes, so their application to “real-life” situations can sometimes be challenging. Due to complexities in cGvHD diagnosis and staging, several studies have shown a lack of adherence to recommendations and inconsistencies in cGvHD evaluation, even among experienced transplant physicians [36, 40–42]. In clinical trials, this can cause patients to be excluded from the analysis post hoc and significantly impact the interpretation of trial results [43]. Therefore, there are increasing efforts to further standardize cGvHD terminology and to improve the quality and precision of the data collected [32–36]. In collaboration with the EBMT Transplantation Complications Working Party and the NIH, Schoemans and colleagues developed an electronic tool, the “eGvHD App,” to assist transplant healthcare professionals with their evaluation of GvHD [33]. Compared to standard practice, the app shows superior GvHD assessment accuracy for both acute and chronic GvHD.
4.2. Complexities in cGvHD Diagnosis and Staging
4.2.1. Late acute and overlap subtype of GvHD
Late acute GvHD (aGvHD) refers to aGvHD features present more than 100 days posttransplant, without evidence of cGvHD. Some studies show higher mortality for patients with late aGvHD compared with classic cGvHD [44, 45], although other studies do not [22, 46]. The incidence of late aGvHD is approximately 11% at 2 years post-alloHCT [47]. With the recognition of the term “late aGvHD,” the incidence of cGvHD has decreased, because many patients previously diagnosed as “chronic,” based on time after transplant, did not meet the diagnostic criteria for cGvHD. Patients with late aGvHD are usually not included in cGvHD treatment studies. The subcategory of the overlap cGvHD is characterized by the presence of aGvHD features in a patient with cGvHD, and has been associated with worse survival [48, 49]. In overlap cGvHD, it might be difficult to determine whether some of the GvHD manifestations (erythematous rash, elevated liver enzymes) are related to acute or chronic GvHD, or to some unrelated process. Although patients with overlap syndrome were originally excluded from many treatment studies, this trend has changed, and those patients are now often considered eligible, although there needs to be detailed documentation of the symptoms to allow for stratification during data analysis.
4.2.2. Differentiating cGvHD from other causes of organ impairment
In patients after alloHCT, there are many possible causes of laboratory abnormalities and organ dysfunction (e.g., infections, medication side effects, iron overload). According to the NIH criteria, if an abnormality is entirely due to a non-cGvHD cause, then that organ should be excluded from cGvHD global severity scoring, although the data should still be captured on the organ-site scoring forms. If cGvHD could at least partly explain organ dysfunction, then the NIH score is used in the global NIH severity scoring without further modification of the score.
4.2.3. Atypical signs and symptoms of cGvHD
Aside from organ systems scored according to the NIH criteria (skin, mouth, eye, gastrointestinal tract, liver, lungs, muscle/joint/fascia, and genitals), there is a variety of additional signs and symptoms, such as ascites, serositis, polymyositis, muscle cramps, nephrotic syndrome, neuropathy, thrombocytopenia, and cardiac involvement, that are associated with cGvHD in 10–15% of cases. If such abnormalities are found and attributed to cGvHD, they should be referred to as “undefined other cGvHD” and recorded in source documents. Many subtle cGvHD manifestations can go undetected if not specifically inquired about. Therefore, patients should be specifically asked about their symptoms and functional impairments at every clinic visit (Fig. 1).
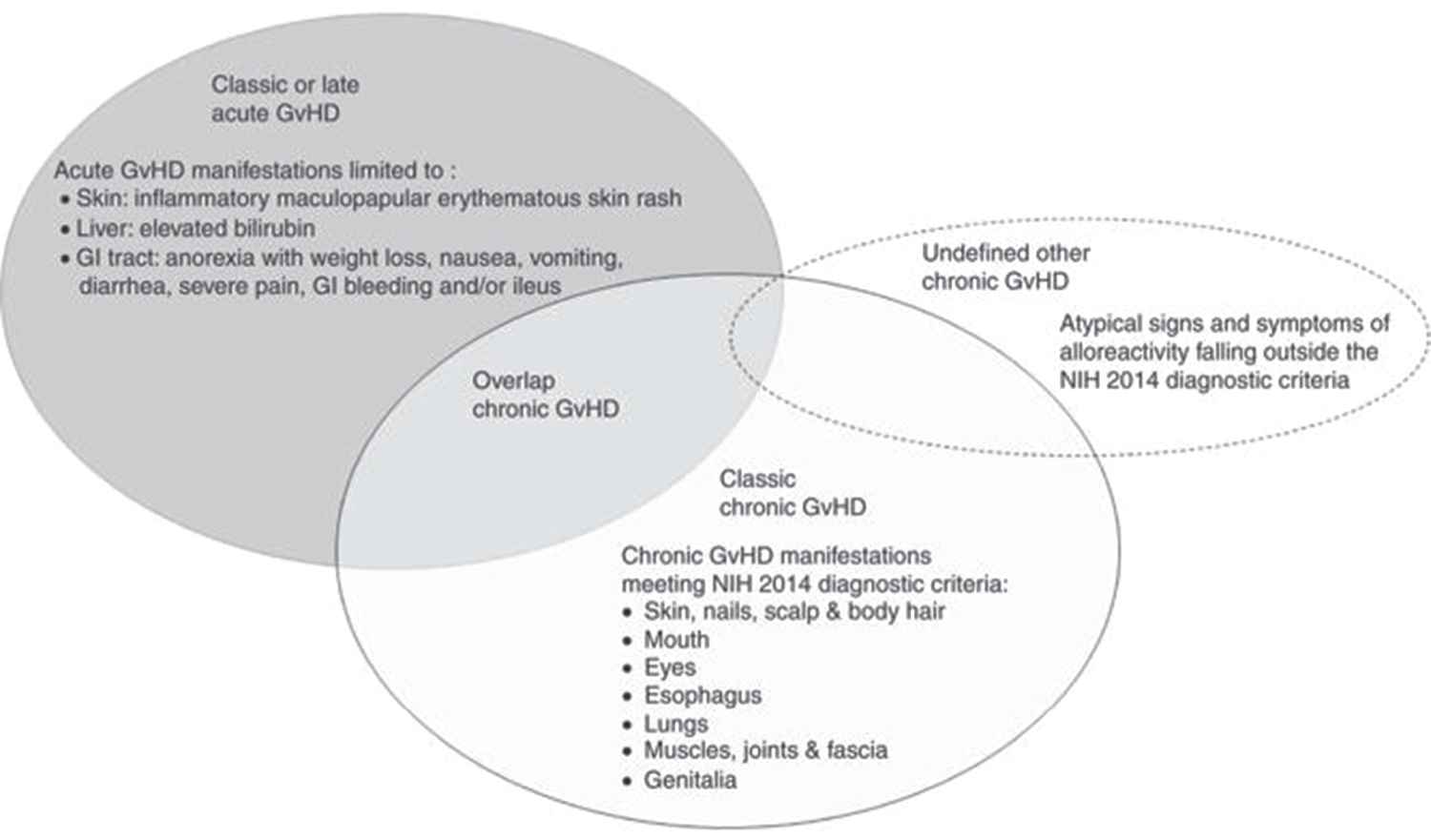
Schematic representation of the types of GvHD.
4.2.4. Third-party input
The evaluation of certain cGvHD manifestations requires a third-party input, such as utilizing spirometry, eye exams, or genital exams. It can be challenging to obtain those on the same visit when cGvHD is assessed by the transplant physician. If expert evaluation (e.g., gynecologist or ophthalmologist) or exact measurements of pulmonary function tests are missing at the time of cGvHD scoring, the NIH score for those organs is utilized based on the patient's symptoms.
5. CHALLENGES IN ASSESSING ELIGIBILITY FOR CLINICAL TRIALS FOR cGvHD
Eligibility criteria will vary depending on whether a study is designed for prophylaxis, treatment of newly diagnosed cGvHD, or treatment of steroid-refractory/dependent cGvHD. In either case, the eligibility criteria should be well defined for the study to ensure the ability to interpret the results.
5.1. Disease Activity and Severity
The presentation of cGvHD can vary from an inflammatory-like syndrome of clinically active disease requiring immediate therapy to a more fibrotic–sclerotic state with fixed, frequently irreversible, deficits which remain after the inflammation resolves. The issue of how to assess fixed deficits in clinical trials should be addressed a priori, because those manifestations likely will not improve with therapy. It is proposed that a definition of cGvHD activity incorporates both the disease manifestations and the need for immunosuppression [34, 35]. Chronic GvHD is considered “clinically active” if the person has worsening inflammatory manifestation, regardless of the use of immunosuppression. After the inflammation resolves, cGvHD manifestations can either resolve or the person may develop fixed, irreversible deficits. Several new terms have been proposed: (1) “controlled cGvHD,” when immunosuppression is still ongoing or has been discontinued for less than 24 weeks, regardless of fixed deficits; (2) “resolved cGvHD,” when immunosuppression has been discontinued for more than 24 weeks and there are no inflammatory signs and no fixed deficits; and (3) “inactive cGvHD,” when immunosuppression is discontinued for more than 24 weeks but fixed deficits persist. Furthermore, the eligibility criteria need to specify the severity of cGvHD for enrollment in the study. The organ severity of cGvHD is scored, by patients' symptoms and functional organ impairment, according to a 4-point scale ranging from 0 (absent) to 3 (most severe). The NIH global cGvHD severity score (mild, moderate, severe) is then derived by combining organ-specific scores [26].
5.2. Response to Steroids
To standardize terminology, transplant physicians “defined” steroid refractoriness and dependence. However, it does not necessarily match the clinical practice, where steroid dosing varies greatly among institutions and practitioners. Steroid-refractory cGvHD is typically defined as progression of cGvHD despite the use of prednisone at ≥1 mg/kg/day for at least 1 week, or no improvement of cGvHD despite the use of prednisone at ≥0.5 mg/kg/day (or 1 mg/kg every other day) for at least 4 weeks. Steroid dependence is defined as the inability to control cGvHD while tapering steroids below 0.25 mg/kg/day in at least two individual attempts separated by at least 8 weeks. Steroid intolerance refers to unacceptable or intolerable side effects from steroids, and it is not equivalent to steroid refractoriness. Prior response to steroids should be clearly defined in the eligibility criteria for clinical trials in cGVHD.
5.3. Prior Lines of Treatment
A line of treatment for cGvHD is defined as an introduction of a systemic treatment (or several treatments, simultaneously), that were not previously used for cGvHD. Dose adjustments to maintain therapeutic levels or to treat cGvHD flare (usually through a temporary increase of steroids) are not considered a new line of therapy. However, restarting the drug that was previously discontinued is considered a new line of therapy. The first-line treatment is defined as the beginning of systemic treatment for cGvHD. Topical organ therapies, ursodiol or fluticasone/azithromycin/montelukast (FAM), are not considered a line of therapy. Oral “nonabsorbable” corticosteroids should be discouraged in cGvHD trials due to their unpredictable systemic effects and drug interactions. The number of prior lines of treatment and the minimum interval time from the most recent change in treatment must be specified in the eligibility criteria and clearly documented in the case report forms. In addition, protocols should specify whether and which prestudy treatments may be continued throughout the study.
5.4. Concomitant Therapy
The protocol should provide guidelines for dosing and adjusting systemic immunosuppression and for starting topical therapies after enrollment. In most trials, one or two temporary steroid pulses, with a rapid taper, are allowed and not considered a treatment failure. It has been shown in many trials, as well as in regular practice, that rigid and fixed steroid taper is not feasible. The study should also specify whether new topical agents are allowed or if their administration is considered a treatment failure. Moreover, the protocol should provide some guidelines for the tapering of systemic immunosuppression in the responding or stable subjects, including for the investigational agent.
5.5. Role of Biomarkers
Although significant progress has been made in the exploration of biomarkers that reliably reflect cGvHD, their specific role in cGvHD trials has not been established [28].
6. CHALLENGES IN ASSESSING TREATMENT RESPONSE IN TRIALS ON cGvHD
The development of standardized response criteria and the identification of robust clinical endpoints for cGvHD trials have been an ongoing challenge, and no gold standard has been established. However, the NIH response criteria were used in the recent FDA approval of ibrutinib for cGvHD after failure of corticosteroids, which established a regulatory standard for future novel drug developments [50]. The treatment “response” generally reflects reducing cGvHD symptoms, decreasing immunosuppression, preventing organ damage, and improving function, HRQOL, and survival. It usually compares disease activity, or “burden,” at specific, predefined, multiple timepoints compared to baseline at the study entry. The “primary endpoint” is the main criterion by which the success of the study drug will be determined and is driven by the phase of the trial design. Endpoints should be based on objective, reliable, and verifiable criteria, standardized and clinically valid measurements should be used, and they should be selected for the ability to demonstrate clinical benefit. Moreover, due to long cGvHD natural history trajectory, the response should not be interpreted under the premise that no response would have occurred in the absence of the investigational drug. The choice of an endpoint will influence the eligibility criteria for the particular study [31]. For example, if the goal of the study is to measure the reduction of symptoms, there should be a minimum burden of symptoms at baseline, necessary to measure clinically meaningful improvement.
The forms for assessing the response should be standardized to allow for better comparison, should be filled out at the baseline and follow-up visits, and should contain adequate details about cGvHD manifestations [29]. It is critical before study entry to document the cGvHD diagnosis and staging on the respective NIH forms [26]. Study coordinators and investigators should conduct real-time data entry and monitoring, to avoid omissions and inconsistencies. It is considered that a 48-hour time frame for such data entry represents a safe practical window for accuracy of “prospective data entry.” Post hoc data abstraction from medical records should not be endorsed due to its notorious inaccuracies.
6.1. Time Frame for Assessing Response
The short-term endpoints, such as dose-limiting toxicities, clinician-assessed or NIH cGvHD response, and patient-reported outcome (PRO), are more suitable for early-phase studies, whereas long-term endpoints, such as survival, therapy failure, and discontinuation of systemic immunosuppression, are needed for late-phase trials. The time frame for assessing the response will also depend on the specific cGvHD manifestations: inflammatory-like manifestations may respond within 4–8 weeks, whereas more fibrotic and sclerotic changes may take 6–12 months to improve/resolve. The 6-month landmark is most commonly used as the primary time point for therapy response assessment. Some severe fibrotic manifestations, such as bronchiolitis obliterans syndrome (BOS), and lacrimal and salivary gland destruction, might be irreversible with current therapies. Several novel endpoints have been proposed over the last few years to improve on cGvHD response assessment as well as to accelerate regulatory approval of new drugs (Table 1).
| Endpoint | Definition | Issues |
|---|---|---|
| Failure-free survival (FFS) | Survival without relapse, nonrelapse mortality (NRM), or new systemic therapy |
|
| Survival without progressive impairment (SWOPI) | Survival without enduring cGvHD-related effects which threaten or compromise physical well-being or function in ways that cannot be easily reversed |
|
| GvHD-free relapse-free survival (GRFS) | Absence of grades 3–4 acute graft-versus-host disease (GvHD), cGvHD requiring systemic therapy, relapse, and death, at one year |
|
| Current GRFS (CGRFS) | Survival without relapse and without active moderate/severe cGvHD at the time of most recent assessment |
|
| Off-immunosuppression relapse-free survival (ISRS) | Survival with withdrawal of all systemic immunosuppression either for therapy or prophylaxis of cGvHD without malignancy relapse or death at 12 months |
|
| Chronic GvHD relapse-free survival (CRFS) | Freedom from development of cGvHD, relapse, or death at 12 months |
|
Composite endpoints for clinical trials.
6.2. Response According to the NIH Criteria
6.2.1. Clinician-assessed response
According to the NIH criteria for cGvHD, categories of response are complete response (CR), partial response (PR), and lack of response (no change, mixed response, progression). To better assess the benefit of PR, there is sometimes a requirement to demonstrate the improvement in the most severe manifestations of cGvHD and improvements across the two categories of severity in a 4-point scale, measured at 6–12 months after the initiation of new treatment [31].
6.2.2. Patient-reported outcome
The incorporation of patient perception of the disease as an endpoint is important in cGvHD clinical trials. PRO measures include patient self-report of cGvHD severity and several different multidimensional HRQOL scales, and may reflect disease activity, adverse effects of drugs, and HRQOL [2, 29, 51–53]. There is increasing evidence supporting the validity of PRO instruments in cGvHD trials, and PROs are frequently used as a secondary endpoint [54, 55]. However, considering that most cGvHD studies are not blinded, the interpretation of interval changes in PROs could be confounded by biases and adverse events and are frequently limited by missing data.
6.3. Composite Endpoints
Composite endpoints acknowledge that the two main risk factors of poor outcome after alloHCT are transplantation-related morbidity/mortality and relapse-related mortality. Efforts to mitigate one can frequently jeopardize the other, as previously mentioned. Therefore, combining these parameters into one composite endpoint is assumed to be a better measure of long-term transplant success.
6.3.1. Failure-free survival
Failure-free survival (FFS) was originally proposed by Inamoto et al. as the absence of relapse, nonrelapse mortality (NRM), or the addition of new systemic immunosuppression for cGvHD [56, 57]. FFS rates at 12 months after initial systemic immunosuppression were at 54%, and after second-line immunosuppression were at 45%. Multivariate analysis identified four risk factors associated with lower FFS in patients receiving initial treatment: onset of initial systemic immunosuppression <12 months posttransplant; patient's age ≥60; severe gastrointestinal, liver, or lung involvement; and Karnofsky score <80%. Risk factors for lower FFS in patients receiving second-line treatment were high-risk disease at transplantation, lower gastrointestinal tract involvement, and severe NIH global score. Subsequent studies examined FFS rates in a multicenter and heterogeneous cohort of patients with both newly diagnosed and existing cGvHD, showing that <50% of patients on systemic immunosuppression will be failure-free survivors at 1 year and <30% of patients will reach 2 years without experiencing failure [55, 58]. Of the variables measured at enrollment, 10 were associated with lower FFS: higher NIH skin score, higher NIH gastrointestinal score, worse range-of-motion score, lower forced vital capacity (%), BOS, worse HRQOL, moderate to severe hepatic dysfunction, absence of treatment for gastric acid, female donor for male recipient, and prior grade II–IV aGvHD. At 6 months, clinician-reported response and NIH-calculated response correlated with FFS, and clinician-reported response predicted overall survival. In addition, a treatment change represented the major cause of failure, as observed in other studies [59,60].
Although FFS can be a useful endpoint in clinical trials, there are several potential limitations associated with it [38]. First, FFS does not give us information about cGvHD-related symptoms, disease activity, the organs involved, and the degree of damage. Thus, studies using FFS as a primary endpoint should measure the response according to the NIH criteria as a secondary endpoint. Second, new treatment decisions are not always driven by a lack of efficacy, but are sometimes led by physician preference, toxicity, price, availability, and convenience, thereby increasing the risk for false-positive and false-negative results. Third, new treatment decisions are subject to bias, making FFS as an endpoint inadequate for regulatory purposes. In the study by Martin et al., FFS with CR/PR at 1 year was associated with clinical benefit, although this occurred in less than 20% of patients [38]. Those patients had lower disease burden, shorter time to the end of systemic treatment, and better survival. The question remains whether the assessment at 6 months after enrollment is too premature to determine the efficacy of the new drug. Overall, FFS coupled with the NIH response serves as a potentially very useful endpoint for phase 2 therapeutic in cGvHD. In addition, reporting the steroid dose at 12 months of therapy with a new agent would increase the specificity and better describe the clinical benefit associated with FFS [56].
6.3.2. Survival without progressive impairment
Survival without progressive impairment (SWOPI) is defined as survival without enduring cGvHD-related effects which threaten or compromise physical well-being or function in ways that cannot be easily reversed [31]. Thus, SWOPI defines the absence of cGvHD progression as a primary measure of success. This endpoint is unaffected by temporary improvement or worsening of reversible cGvHD manifestations, and it could be of relevance in patients with advanced cGvHD despite multiple therapies. The absence of cGvHD progression and organ damage without treatment-related toxicity would potentially be valuable measures of success, even on continuous therapy. The investigational agent with a good SWOPI score could potentially replace steroids for long-term use. However, methods to measure SWOPI are not fully developed and there are many potential confounders.
6.3.3. GvHD-free relapse-free survival and current GvHD-free relapse-free survival
A new composite endpoint of GvHD-free relapse-free survival (GRFS) was proposed, measuring a cure without ongoing morbidity [61]. It is defined as the absence of: grade 3–4 aGvHD, cGvHD requiring systemic therapy, relapse, or death, at one year. In a retrospective study, Holtan et al. found that only approximately one-third of patients survived one year without experiencing a GRFS-defining event [61]. In addition, GRFS differs significantly based on patient age, disease risk, and donor type [62, 63]. The two main concerns associated with GRFS are (1) it only measures time to the first event, thus underestimating the success of alloHCT and (2) it treats grade 3–4 aGvHD and cGvHD, requiring immunosuppression as “fixed” deficits. Although grade 3–4 aGvHD can be associated with significant morbidity and mortality, treating it as a separate event is not necessary, because it will either resolve or convert to either cGvHD or death. To address these issues, the new composite endpoint was proposed and termed the “current GRFS” (CGRFS), defined as survival without relapse and without active moderate/severe cGvHD at the time of the most recent assessment [64]. CGRFS has the potential to be a better surrogate of transplant success and is not impacted by age, donor type, regimen, or stem cell source. In a study by Solomon et al., the CGRFS decreased continuously after transplant, accounting for 23%, 14%, 7%, and 4%, respectively, in the first 4 years [64]. At 1-year posttransplant, the estimated disease-free survival, CGRFS, and conventional GRFS were 68%, 45%, and 33%, respectively, and at 4 years they were 52%, 49%, and 22%, respectively (Fig. 2). However, this study was limited to a single center, thus further validation and refinement of CGRFS is required through multicenter studies.
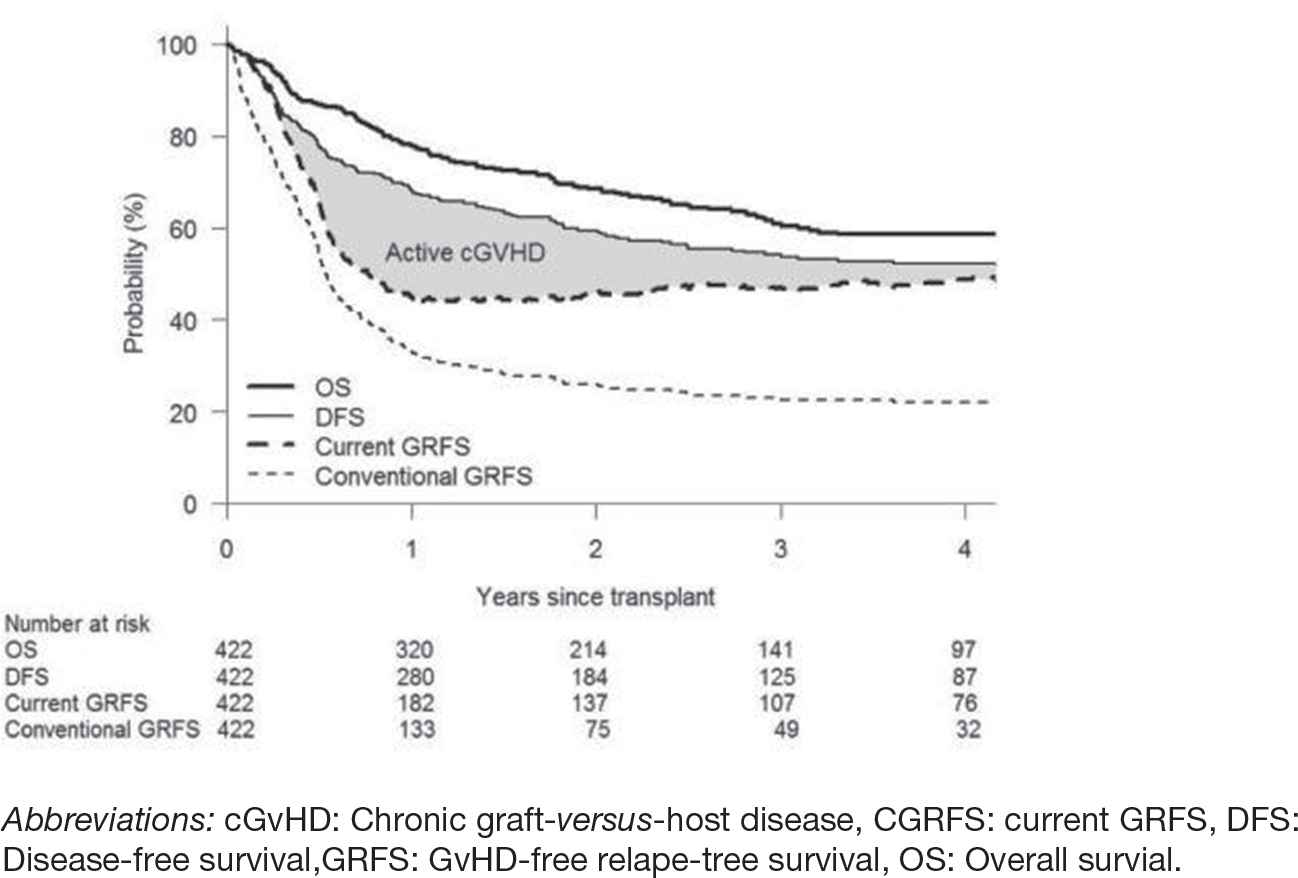
Comparison of OS, DFS, GRFS, and CGRFS.
6.3.4. Off-immunosuppression relapse-free survival and cGvHD relapse-free survival
Several novel composite endpoints were explored in the study comparing GvHD prevention strategies from six single institutions with a large multicenter database as a control [65]. Immunosuppression relapse-free survival (ISRS) is defined as survival with the withdrawal of all systemic immunosuppression for cGvHD, either for therapy or prophylaxis, without malignancy, relapse, or death at 12 months. cGvHD relapse-free survival (CRFS) is defined as freedom from the development of cGvHD, relapse, or death at 12 months. A recent study by Lee et al. showed that for patients with cGvHD who start initial systemic immunosuppression, there is only a 32% chance that they will be alive, in remission, and off immunosuppression (e.g., ISRS) 5 years after cGvHD diagnosis [66]. Use of these composite endpoints in prevention trials could increase the precision of outcome analysis in regard to GvHD, relapse, and survival. Subsequently, two multicenter trials addressing GvHD prevention were initiated through the Clinical Trials Network (PROGRES 1 and 2), further exploring those composite endpoints, including GRFS.
6.4. Controlled Trial Design
In controlled trials, stratified randomization decreases the probability of imbalance between groups, although such trials require large sample sizes to assess the statistical difference and to determine the true treatment effect. Controlled trials might pose an ethical dilemma for investigators, as well as patients, regarding enrollment in a study testing a new drug against an already-established conventional therapy or the use of placebo. A crossover design could be used to overcome this limitation and could allow all patients exposure to the investigational agent.
7. CONCLUSIONS
Various clinical presentations and the heterogeneity of organ involvement in cGvHD, coupled with the absence of reliable biomarkers of disease activity, delay progress in the discovery of new treatment modalities. There is an urgent need for faster development and implementation of well-designed therapeutic trials, both in newly diagnosed and steroid-refractory cGvHD. Novel strategies to reduce or eliminate the need for long-term corticosteroids are of primary interest. The development of better preemptive treatment approaches, which would serve as prophylaxis or interfere with the progression of evolving cGvHD, has legitimate potential to advance clinical practice. This would lead to reduced disability, improved patient satisfaction, better HRQOL, and an ultimately lower cost. The transplant community needs to advocate for support of these cGvHD studies. Only with that level of dedication and joint effort will progress be made in this disorder.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
ACKNOWLEDGMENT
We apologize to many colleagues worldwide whose excellent manuscripts we were unable to cite in this review because of space constraints. We thank Ms. Glenn Floyd for editorial assistance.
REFERENCES
Cite this article
TY - JOUR AU - Iskra Pusic AU - Steven Z. Pavletic PY - 2019 DA - 2019/03/18 TI - Challenges in Conducting Studies in Chronic Graft-versus-Host Disease JO - Clinical Hematology International SP - 36 EP - 44 VL - 1 IS - 1 SN - 2590-0048 UR - https://doi.org/10.2991/chi.d.190314.001 DO - 10.2991/chi.d.190314.001 ID - Pusic2019 ER -