
Management of Sickle Cell Intrahepatic Cholestasis: An Argument in Favor of Automated Exchange Transfusion
- DOI
- 10.2991/chi.d.190630.001How to use a DOI?
- Keywords
- Sickle cell hepatopathy; Sickle cell disease; Apheresis; Red blood cell exchange; Exchange transfusion
- Abstract
Sickle cell disease patients are commonly treated at transfusion medicine services, and understanding of the hepatic manifestations of the disease is key for optimal management, specifically, in individuals presenting with sickle cell intrahepatic cholestasis (SCIC). SCIC represents a rare, severe hepatic crisis wherein sinusoidal red cell sickling leads to massive hepatocyte dysfunction and cholestatic laboratory findings. Acute SCIC is defined by abdominal pain with progressive hepatic injury associated with hyperbilirubinemia, renal failure, encephalopathy, and coagulopathy. Patients are generally managed with red blood cell exchange transfusion (RBCEx), when available, as this is a potentially fatal condition. Simple transfusion may be utilized in resource-poor environment or when patients refuse RBCEx. As less than 50 adult cases have been described in the literature, many of them with limited follow-up, randomized clinical trials comparing RBCEx with other treatments are currently unfeasible. Likewise, a chronic form exists, but is less well characterized, and is associated with persistent bilirubinemia and a variable course in terms of progressive hepatic disease. We undertake a brief review of the literature and discuss two cases of SCIC managed with RBCEx at our institution.
- Copyright
- © 2019 International Academy for Clinical Hematology. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
Sickle cell disease (SCD) is the most common hemoglobinopathy, and there are approximately 100,000 patients in the United States with the disease [1,2]. This results in a major impact on healthcare, with between 68 and 75,000 hospitalizations a year, and approximately 100,000 emergency department visits, with an estimated total cost of care of $2.4 billion [3]. The rate of hepatic complications of SCD is unknown, but the prevalence of hepatic dysfunction is estimated at 10%, accounting for 0.8% of all-cause mortality [4,5]. Sickle cell hepatopathy is a broad term encompassing numerous changes seen in SCD which may be caused by physiologic sickling and sequestration, iron overload, various hepatidides, or other multifactorial causes.
Acute sickle cell intrahepatic cholestasis (SCIC) is a severe complication of sickle cell hepatopathy, which occurs due to sickling within the hepatic circulation and subsequent ischemia [6]. Histologically, SCIC is characterized by sickled erythrocytes within sinusoids, ballooning hepatocytes, and dilated canaliculi with bile plugs [7,8]. Chou et al. defines SCIC as, “the sudden onset of right upper quadrant abdominal pain, significantly elevated transaminases (>1000 mg/dL), severe hyperbilirubinemia (total serum bilirubin often >50 mg/dL), coagulopathy, hepatomegaly, renal insufficiency, and acute liver failure in severe cases” [6].
SCIC can present acutely or recurrently as chronic process which may give rise to overt liver failure [5,7,9]. To date, seven cases of chronic SCIC have been described in adults, and the entity has no fixed clinical or histologic criteria [5,10–14]. Symptomology is defined by pronounced hyperbilirubinemia with variable associated symptoms and intermittent exacerbating episodes of acute SCIC [5,7,9].
Though SCD was first reported in 1910, the description of hepatic manifestations was limited, with most publications focusing on hepatomegaly and elevated bilirubin [15,16]. In 1953, a case of SCIC was described by Green et al. [5,16]. The patient presented with severe abdominal pain, progressive jaundice, and bleeding abnormalities, and would go on to expire secondary to hepatic coma [16]. An autopsy was performed, and the liver demonstrated histologic evidence of sickling and cholestasis [16]. Early cases of SCIC in adults treated with supportive therapy, such as simple transfusion and peritoneal dialysis, were associated with 88% mortality (Table 1), compared to pediatric cases where survival was more commonly reported [17–19]. In 1980, use of partial red cell exchange in the management of an adult SCD patient with SCIC, resulting in survival, was first described [18].
| Year of Publication | Age | Genotype | Max. Total Bilirubin (mg/dL) | Exchange | Reference |
|---|---|---|---|---|---|
| 1953 | 33 | HbSS | 112 | − | [16] |
| 1957 | 27 | HbSS | 17 | − | [8] |
| 1964 | 24 | HbSS | 100 | − | [8] |
| 1965 | 23 | HbSS | 81 | − | [27] |
| 1965 | 22 | HbSS | 89 | − | [27] |
| 1965 | 20 | HbSS | 61 | − | [27] |
| 1977 | 44 | HbSS | 98 | − | [8,27] |
| 2005 | 27 | HbSS | 80 | + | [22,27] |
| 2006 | 48 | HbS/β | 60 | + | [27] |
| 2015 | 45–49 | HbSS | 59 | − | [38] |
| 2017 | 38 | HbSS | 16 | + | [28] |
Clinical characteristics of adult patients (>18 years of age) who died due to acute SCIC.
In the intervening years, significant advances have been made in the management of SCD, and supportive management utilized in the treatment of SCIC has progressed with improvements in dialysis, critical care medicine, and solid organ transplant [20]. Reported cases of SCIC have been routinely managed with red blood cell exchange transfusion (RBCEx). Of cases reported since 1980, two patients survived without the use of RBCEx; however, due to the rarity and risk of mortality, a direct comparison through randomized controlled trials (RCT) of simple transfusion versus RBCEx has not been performed [21–24].
Management of chronic SCIC is less extensively characterized, due to a paucity of reported clinical cases, and to the clinical overlap with other progressive liver disease occurring in SCD patients, such as autoimmune hepatitis, iron overload, or viral hepatitis. Many patients are managed with RBCEx at regular intervals and for acute episodes. To date, two reported patients with chronic SCIC have been successfully managed with orthotopic liver transplant (OLT) [5,12]. The American Society for Apheresis (ASFA) guidelines pertaining to RBCEx recommendations, and management of SCIC remain quite [25]. Thus, evaluation must be made on a case by case basis. However, the significant risk of acute mortality or requirement for OLT in chronic disease supports the use of RBCEx therapy, if available.
2. METHODS
A chart review was performed of the Vanderbilt University Medical Center (VUMC) electronic medical record (Starpanel, HealthIT@Vanderbilt, Nashville, TN). A natural language search of the VUMC laboratory information system (CoPath, Sunquest, Tuscon, AZ) for cases of acute SCIC was performed. All liver biopsy specimens, estimated approximately 25,000 cases, and autopsy cases, estimated approximately 3000 cases total, from 1999 to present were queried. No institutional review board approval was required, as only autopsy materials were reviewed and all other records were deidentified.
3. PRESENTATION OF ACUTE SCIC
Acute SCIC presentations vary, but manifestations are characterized by progressive liver failure. Patients characteristically have elevated total bilirubin, variable changes in liver function tests, and upper abdominal pain. Many patients without underlying hepatic disease have significant improvement with RBCEx therapy, though mortality has been described despite intervention [26–28]. We present a case report of a patient we saw in our clinic.
3.1. Case 1
A 26-year-old African American male with a history of SCD, HbSS genotype, presented with a 2-day history of worsening epigastric abdominal pain. On exam, the patient was jaundiced, had abdominal tenderness, but no evidence of hepatosplenomegaly. Total bilirubin was elevated to 24.9 mg/dL (reference range 0.2–1.2 mg/dL) along with aspartate transaminase (AST) 176 units/L (reference range 5–40 units/L) and alanine transaminase (ALT) 108 units/L (reference range 0–55 units/L). Hepatitis B and C as well as HIV testing was negative. An MRI demonstrated no hepatomegaly and an atrophic spleen consistent with chronic SCD. Right upper quadrant ultrasound was notable for cholelithiasis and he underwent cholecystectomy and cholangiopancreatography for removal of a common bile duct stone.
On day 10 in hospital, he continued to have increased total bilirubin with a peak of 80 mg/dL and also developed a prolonged prothrombin time (PT), 18.9 seconds (reference range 11–14.6 seconds). A presumptive diagnosis of SCIC was made, and the patient underwent RBCEx with a goal of 20% HbS utilizing six units of crossmatched-compatible red blood cells (RBC) which were C, E, and Kell phenotypically matched and SICKLEDEX® (Streck, Omaha, NE) negative on the COBE Spectra Apheresis System (Terumo BCT, Lakewood, CO). His hemoglobin S level decreased from 44.4% to 17.8%. His laboratory markers improved, and the patient was discharged (Table 2, Figure 1). At 2 years after discharge, the patient has returned to his clinical and laboratory baseline without development of chronic SCIC.
| Patients with SCIC |
|||||
|---|---|---|---|---|---|
| Patient 1 |
Patient 2 |
||||
| Parameter | Reference Range | Value prior to RBCEx | Value at time of discharge | Value prior to RBCEx | Value at time of discharge |
| Hemoglobin S percentage | 44.4% | 38.5% | 44.1% | 22.6% | |
| Total bilirubin | 0.2–1.2 mg/dL | 80 mg/dL | 31.8 mg/dL | 59.1 mg/dL | 44.1 mg/dL |
| Alanine aminotransferase (ALT) | 0–55 mg/dL | 160 mg/dL | 201 mg/dL | 42 mg/dL | 53 mg/dL |
| Aspartate aminotransferase (AST) | 4–40 mg/dL | 175 mg/dL | 184 mg/dL | 160 mg/dL | 161 mg/dL |
| PT | 11–14.6 seconds | 18.9 seconds | 13.3 seconds | 24.5 seconds | 17.3 seconds |
| BUN | 7–21 mg/dL | 16 mg/dL | 5 mg/dL | 36 mg/dL | 21 mg/dL |
| Creatinine | 0.57–1.25 mg/dL | 1.03 mg/dL | < 0.2 mg/dL | 4.35 mg/dL | 0.4 mg/dL |
Abbreviations: RBCEx: automated red blood cell exchange.
Laboratory values of two adult patients with SCIC managed with RBCEx.
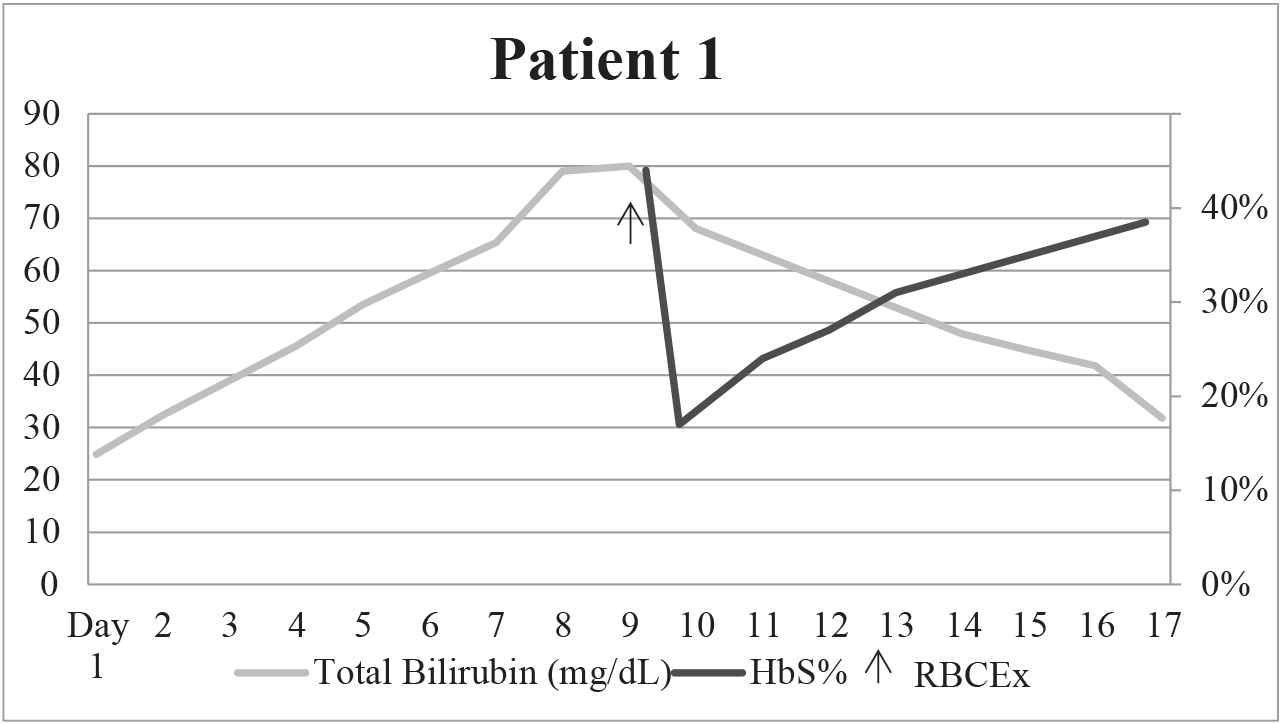
Graphical depiction of HbS% and total bilirubin over time before and after RBCEx for patient 1.
4. PRESENTATION OF CHRONIC SCIC WITH ACUTE EXACERBATION
Chronic SCIC is less well characterized, but patients routinely have variable baseline liver dysfunction with intermittent exacerbation. Few cases have been previously described. Patients present with chronically elevated bilirubin, as is characteristic of SCD, likely due to baseline hemolysis as well as liver dysfunction [5,10–14]. We herein present a case report of a patient we saw in routine practice and followed thereafter.
4.1. Case 2
A 45-year-old African American female with HbS/β0 thalassemia, autoimmune hepatitis, and iron overload presented with fatigue. Her past surgical history was notable for cholecystectomy for cholelithiasis. She was admitted for hepatic encephalopathy and renal failure. On physical exam, the patient was icteric and somnolent, without evidence of hepatosplenomegaly. The total bilirubin was elevated to 59.1 mg/dL (reference range 0.2–1.2 mg/dL) along with elevations in blood urea nitrogen (BUN) and creatinine and a prolonged prothrombin time (Table 2). Hepatitis B and C, HSV-1 and 2, and HIV testing was negative. Ultrasonography demonstrated no gallbladder, an infarcted spleen, and no hepatomegaly. The patient was admitted to the medical intensive care unit.
The patient initially received two simple transfusions of red blood cells for anemia, 7.8 g/dL hemoglobin (reference range 11.8–16), and presumed SCIC. This lowered the patient's HbS from 44.1% to 27.9%. However, her clinical status did not improve, and the medical team proceeded with RBCEx. The patient underwent this procedure with a goal of 25%HbS, utilizing three units of PRBCs which were crossmatch-compatible with PRBCs that were C, E, and Kell phenotypically matched and SICKLEDEX® negative on the COBE Spectra Apheresis System. Her HbS was reduced from 27.9.1% to 17.1%. Her laboratory parameters improved following RBCEx (Table 2, Figure 2). The patient stabilized clinically with continuous renal replacement therapy dialysis and supportive therapy, and was discharged. She managed as an outpatient with monthly partial exchange, and continued to have elevated total bilirubin ranging from 14.4 to 32.9 mg/dL and HbS ranging from 50.8% to 74.7%. She had four subsequent hospital admissions, two of which required RBCEx. During her second admission for RBCEx at 14 months follow-up, the patient expired due to multisystem organ failure in a setting of pulmonary hypertension and end-stage liver and kidney disease. A postmortem analysis was not conducted.
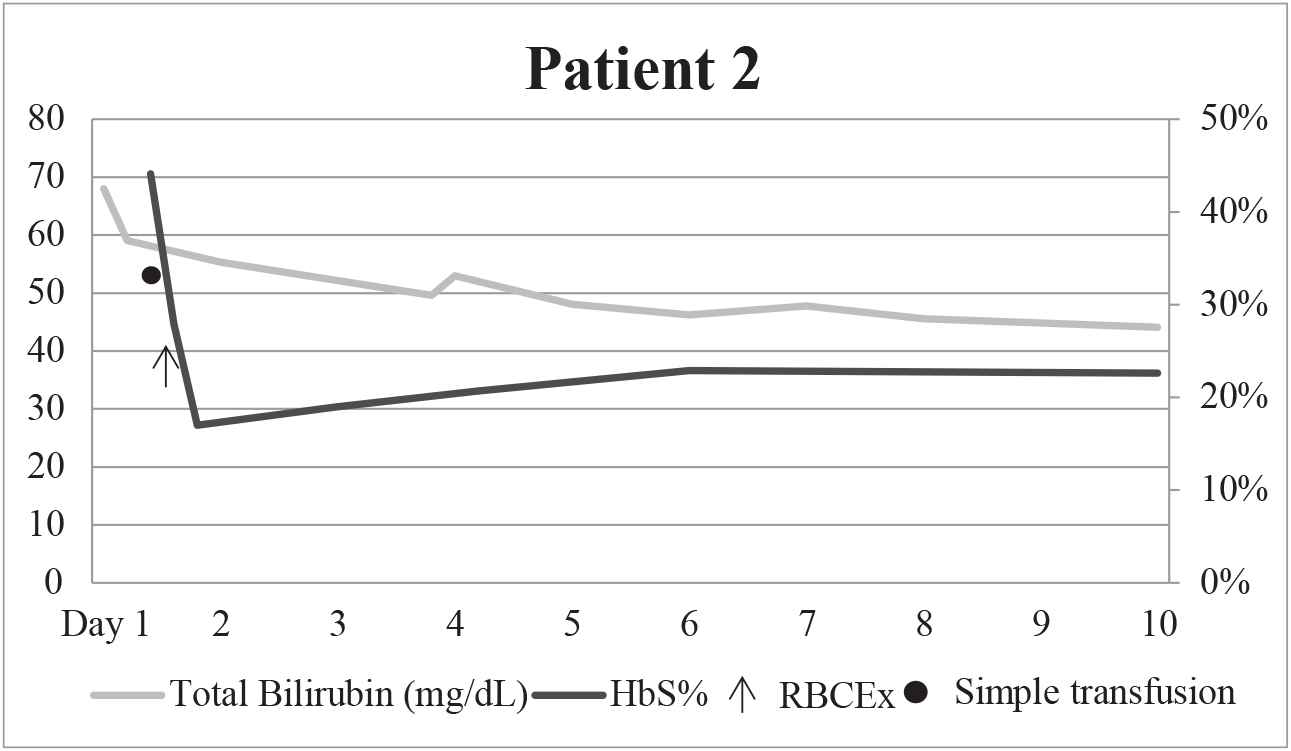
Graphical depiction of HbS% and total bilirubin over time before and after RBCEx for patient 2.
5. RESULTS OF LABORATORY INFORMATION SYSTEM QUERY
No liver biopsy specimens with acute SCIC were identified. A single autopsy case was signed out as acute SCIC; however, upon review of the materials and medical records, this more likely represented another etiology of hepatic failure in an SCD patient, as there was no pronounced hyperbilirubinemia reported or significant sickling or cholestasis in the submitted liver sections
6. DISCUSSION
SCIC can be a potentially fatal manifestation of SCD, therefore appropriate identification and management in consultation with transfusion medicine services is essential. The cases we report here represent a spectrum of the adult patients who present with SCIC with varying baseline liver function. Young patients with normal hepatic reserve who present acutely with SCIC, tend to experience a full recovery, as exemplified by patient 1. The diagnosis was delayed while other medical and surgical etiologies of right upper quadrant pain were evaluated, as may be the case for other SCIC patients, because the differential diagnosis for abdominal pain in patients with SCD is broad. Despite the prolonged interval prior to receiving exchange therapy, the patient had otherwise normal liver and kidney function, and no chronic diseases outside of SCD. As a result, the patient responded well to RBCEx, had a rapid recovery, and has demonstrated no further hepatic or renal symptomology at follow-up 2 years later.
Conversely, in older patients with underlying liver disease, SCIC exacerbations can be more deleterious. Though SCIC was rapidly identified in patient 2, and she received simple transfusion while she was being evaluated for RBCEx, her acute symptoms did not improve immediately following the procedure. She continued to receive prophylactic partial red cell exchange every 6 weeks, and her total bilirubin remained elevated. Despite preventative as well as emergent exchange therapy, she continued to have elevated total bilirubin and, at 14 months follow-up, the patient expired due to progressive organ failure.
There are many diagnostic pitfalls in the evaluation of SCIC. Acute upper abdominal pain is associated with a number of diagnoses, and interpreting bilirubin levels may be challenging in SCD patients in whom they may be chronically elevated [6]. The diagnosis may be rendered via liver biopsy; however, this procedure in acute sickle cell hepatopathy has been associated with a 36% risk of serious hemorrhage and a mortality of 28% [29]. Additionally, strict histologic criteria for diagnosis are not defined. Therefore, the diagnosis is dependent on clinical presentation and laboratory value perturbations.
Issues with diagnosis carry over into the medical literature with numerous reports using mixed descriptors for SCIC. SCIC is a specific clinical entity with features that are distinct from hepatic sequestration, and overlap with the latter may contribute to underreporting [30]. The current ASFA guidelines regard the two as a single entity in terms of treatment which, given their similar pathogenesis and outcomes, seems reasonable [25].
From 1953 to the present, we reviewed all published reports of adult patients diagnosed with SCIC. The first description of exchange therapy occurred in 1980. However, prior to Sheehy et al., there was 88% mortality in patients over 18 years of age presenting with SCIC [18]. From 1980 onwards, 22 cases of acute SCIC in adults have been reported, with 86% of patients undergoing exchange transfusion with a survival rate of 84% (Table 3). Two of three who did not undergo exchange survived (Table 3).
| Year of Publication | Age | Genotype | Max. Total Bilirubin (mg/dl) | Exchange | Reference |
|---|---|---|---|---|---|
| 1960 | 23 | HbSS | 45 | − | [17] |
| 1980 | 21 | HbSS | 146 | Partial | [18,27] |
| 1986 | 35 | HbSS | 75 | Partial | [39] |
| 1995 | 29 | HbSS | 106 | + | [25,40] |
| 1996 | 36 | HbS/β | 47 | − | [21,27] |
| 2002 | 37 | HbSS | 61 | + | [27,41] |
| 2002 | 22 | HbS/β | 55 | + with OLT | [10,27] |
| 2003 | 21 | HbSS | 48 | + | [11,22,42] |
| 2004 | 40 | HbS/β | 55 | + | [22,43] |
| 2011 | 41 | HbSS | 59 | + | [22,44] |
| 2011 | 21 | HbSS | 14 | + | [22,45] |
| 2014 | 48 | HbSS | 79 | − | [22] |
| 2014 | 37 | HbS/β | 47 | + | [12] |
| 2015 | 31 | HbSS | 50 | + | [46] |
| 2019 | 45 | HbS/ |
68 | + | |
| 2019 | 26 | HbSS | 80 | + | |
| 2017 | 32 | HbS/β | 7 | + | [13] |
| 2017 | 54 | HbS/β | 12.4 | + | [13] |
| 2018 | 29 | HbSS | 77 | + with OLT | [31] |
Source: Data compiled and presented with permission from Morrow and McKenzie [39], Costa et al. [27], Hosiriluck et al. [22], Vlachaki et al. [12], Gardner et al. [5], Malik et al. [46], D'Ambrosio et al. [13], and Kwun Lui et al. [31]. The two patients presented in this article are included for completeness.
Abbreviations: Hb: hemoglobin; OLT: orthotopic liver transplant.
Clinical characteristics of adult patients (>18 years of age) with survival and follow-up after acute SCIC.
The impact of SCIC is evident in patients who have died in spite of RBCEx [26–28]. These cases highlight the unpredictable severity of SCIC, and suggest that patients with more severe liver disease or acute injury are less amenable to RBCEx therapy [26–28]. OLT may be utilized in patients recalcitrant to RBCEx, though it is not always successful [26,31].
Management of chronic SCIC with OLT has been efficacious in two patients with recurrent SCIC [5,10]. Gardner et al. formulated an algorithm for OLT in patients with SCIC. Criteria for SCIC were hyperbilirubinemia >200 μmol/L or >12 mg/dL, exclusion of hyperhemolysis or other liver disease, and predominantly conjugated hyperbilirubunimia with minimal liver damage. Chronicity defines management: for the first episode one should consider RBCEx; for the second, regular RBCEx, and for progressive hepatopathy; RBCEx program and evaluation for OLT are recommended.
Shah et al. reviewed sickle cell hepatopathy including a report of OLT in this population [32]. They describe 18 cases of SCD managed with OLT. The etiology of liver disease in these SCD patients was not included, but outcomes varied significantly among the reviewed cases, with some series having up to 100% mortality in the posttransplant period. Though success was described in roughly half the cases, OLT was associated with significant mortality and morbidity in the posttransplant period [32]. Likewise, recurrent disease or other manifestation of sickle cell hepatopathy may occur in the transplanted liver [32].
Other means of SCD management must also be considered such as routine follow-up with a hematologist. Hydroxyurea therapy has been well-established in SCD, and may be effective in preventing acute exacerbations of SCIC [12]. Another consideration may be allogeneic stem cell transplant (SCT), the only currently available definitive cure for SCD. Though progressive liver disease is not a currently recognized indication, efficacy has been demonstrated. [33–36]. SCT is likewise associated with the risk of graft versus host disease, as well as hepatic veno-oclusive disease [37]. Dual SCT/OLT has been described in other disease processes, and may be an avenue explored in SCD patients in the coming years [1].
The most comprehensive clinical and literature-based review of sickle cell hepatopathy to date was performed by Ahn et al., who evaluated both pediatric and adult cases from 1953 to 2002, as well as a 20-year review of pediatric patients with sickle cell hepatopathy at their institution [19]. Cases were grouped by presentation, as mild and severe. Those with severe presentation meeting the criteria for SCIC were found to have an overall mortality of 64%, being 92% without exchange therapy and 22% when receiving exchange. The authors described RBCEx as the “only effective therapy” for patients with severe presentation, and recommended a clinical workup and close monitoring with prompt RBCEx for evidence of severe hepatic dysfunction.
A natural language search of the pathology results of approximately 28,000 cases at our institution yielded scant results. No liver biopsy specimens with acute SCIC were identified, which is likely due to the high mortality associated with the procedure. The single misdiagnosed autopsy case is informative, as SCIC continues to represent a diagnostic challenge. As VUMC is a large referral center for SCD patients, the lack of biopsy or autopsy in proven SCIC cases supports that this is a rare manifestation.
As SCIC prevalence is low, conclusions must be drawn from case series or reports and, unfortunately, many early descriptions are hampered by low-quality data. Additionally, prior characterizations suffer from a lack of a diagnostic gold standard for SCIC. However, in 95% of published cases, consideration or performance of RBCEx are mentioned. Personalized consultation for each case of SCIC is advised, and patients who appear to be developing liver failure, especially those with ongoing liver disease, should be effectively triaged for RBCEx.
Despite outcomes that would support the use of exchange transfusion, the procedure remains a weak recommendation from professional apheresis committees, due to limited evidence. However, transfusion services must carefully evaluate these highly acute patients, due to the risk of mortality [23]. We believe that the ASFA recommendation overlooks published evidence and may be indicative of inappropriate patient management. To date, outside of a limited number of case reports, no compelling evidence has been presented to argue against RBCEx in the management of SCIC. As such, despite limited data, we suggest using RBCEx as a first-line management, due to successful outcomes in the available literature. Few studies have demonstrated success with OLT, though this is significantly less well characterized and, as such, should be considered on a case by case basis. In the acute setting, we suggest a treatment algorithm with simple transfusion in resource-poor environments as a bridge to RBCEx, immediate RBCEx if available, and consideration for OLT for progressive or recurrent symptoms.
CONFLICT OF INTEREST
The authors declare no competing financial interests. There is no conflict of interest.
Footnotes
REFERENCES
Cite this article
TY - JOUR AU - Brian D. Adkins AU - Bipin N. Savani AU - Garrett S. Booth PY - 2019 DA - 2019/09/01 TI - Management of Sickle Cell Intrahepatic Cholestasis: An Argument in Favor of Automated Exchange Transfusion JO - Clinical Hematology International SP - 127 EP - 133 VL - 1 IS - 3 SN - 2590-0048 UR - https://doi.org/10.2991/chi.d.190630.001 DO - 10.2991/chi.d.190630.001 ID - Adkins2019 ER -