
History of Acute Promyelocytic Leukemia
 , Eva Barragán2, 3
, Eva Barragán2, 3- DOI
- 10.2991/chi.k.210703.001How to use a DOI?
- Keywords
- Acute promeloctic leukemia; arsenic trioxide; all-trans retinoic acid
- Abstract
In this article, we discuss the history of acute promyelocytic leukemia (APL) from the pre-therapeutic era, which began after its recognition by Hillestad in 1947 as a nosological entity, to the present day. It is a paradigmatic history that has transformed the “most malignant leukemia form” into the most curable one. The identification of a balanced reciprocal translocation between chromosomes 15 and 17, resulting in fusion between the promyelocytic leukemia gene and the retinoic acid receptor alpha, has been crucial in understanding the mechanisms of leukemogenesis, and responsible for the peculiar response to targeted therapy with all-trans retinoic acid (ATRA) and arsenic trioxide (ATO). We review the milestones that marked successive therapeutic advances, beginning with the introduction of the first successful chemotherapy in the early 1970s, followed by a subsequent incorporation of ATRA and ATO in the late 1980s and early 1990s which have revolutionized the treatment of this disease. Over the past two decades, treatment optimization has relied on the combination of ATRA, ATO, and chemotherapy according to risk-adapted approaches, which together with improvements in supportive therapy have paved the way for cure for most patients with APL.
- Copyright
- © 2021 International Academy for Clinical Hematology. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
Paraphrasing Bernard [1], the exciting history of acute promyelocytic leukemia (APL) is marked by a series of efforts to combat existing dogmas in cancer. In recent years, APL has become a paradigm that has opened new paths for the treatment of not only other leukemias, but many other forms of cancer.
In the history of APL, we can schematically recognize the following periods: (1) The pre-therapeutic era (1940–1973), in which the promyelocytic cell achieved full recognition for the first time, followed by APL being considered as a unique nosological entity some years later; (2) The chemotherapy era (1973–1988), in which important advances were also made in the biological and genetic characterization of this peculiar leukemia; (3) The modern era (1988–present), in which the advent of all-trans retinoic acid (ATRA) and later arsenic trioxide (ATO) have revolutionized the treatment of APL. During this period, progress has been remarkable through the implementation of strategies that have optimized the combination of the three fundamental agents in this disease: ATRA, ATO and chemotherapy. Figure 1 shows the most important milestones that have marked the history of APL and paved the way for the cure of most patients with this disease.
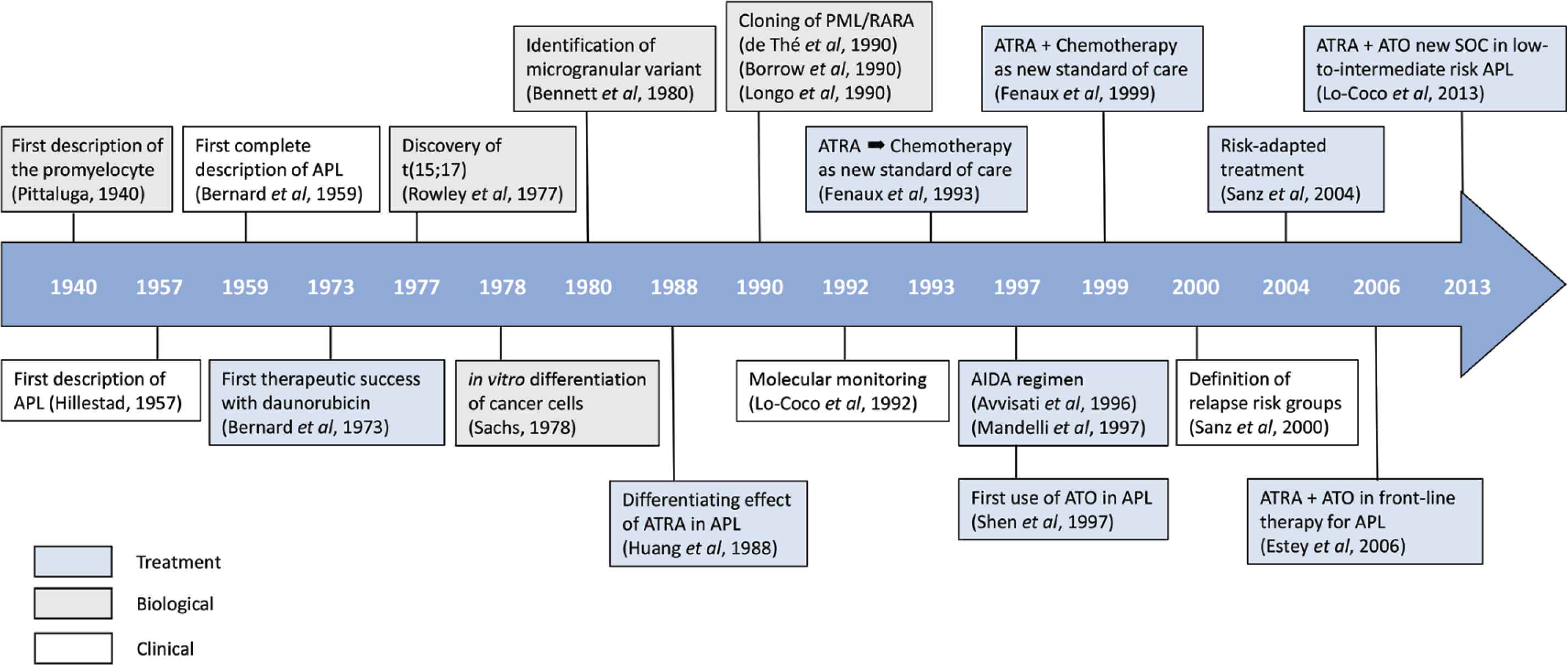
Main milestones in the history of acute promyelocytic leukemia.
2. THE PRE-THERAPEUTIC ERA
2.1. Recognition of the Promyelocyte as a Cellular Entity and APL as a Nosological Entity
Although the term promyelocyte had already been used years before by Pappenheim [2], it was not until 1940 that a permanent recognition of this cell occurred after the excellent description made by the Italian-Spanish Pittaluga [3].
After that, the next milestone was the recognition of APL as a nosological entity. The merit must be attributed to the Norwegian Hillestad [4], who, in a short and precise article, described the disease in three patients characterized by “a very rapid fatal course of only a few weeks duration”, a leukocyte picture dominated by the presence of promyelocytes, and a tendency to severe bleeding due to fibrinolysis and thrombocytopenia. Hillestad also observed a normal erythrocyte sedimentation rate, probably caused by a low plasma fibrinogen concentration. His conclusion was, therefore, that “APL appears to be the most malignant form of acute leukemia.” He was right, referring to the natural history of this disease, since at that time there was a lack of any effective form of treatment. Fortunately, as we will discuss later, the natural history of APL would begin to change with the first therapeutic successes reported by Bernard et al. [5]. Returning to Hillestad’s study, he should also be credited with introducing the term by which this peculiar form of acute myeloid leukemia (AML) has been known since then, when he said “a logical name for this type of leukemia is acute promyelocytic leukemia”. Almost simultaneously, a relationship between promyelocytic proliferation and a hemorrhagic syndrome in patients with leukemia was found by Bernard’s group in Paris [6]. A couple of years later, this group described in more detail the features of APL in a series of 20 patients [7]. They confirmed the hyperacute course of APL, with abundant mucocutaneous hemorrhage that led to death due to cerebral hemorrhage in a high proportion of patients. The malignant cells in the blood and bone marrow of patients with APL had a characteristic morphology along with severe fibrinolysis and hypofibrinogenemia. Typically, malignant cells were much more abundant in bone marrow than in peripheral blood, where white blood cell counts were typically low. These malignant cells were described as abnormal promyelocytes with a characteristic pattern of heavy granulation, often reniform or bilobed immature nucleus, and a large cytoplasm with abundant azurophilic granulation, sometimes nearly covering the nucleus. Many of these cells contained characteristic formations in the cytoplasm, called Auer rods, which often formed bundles (“faggots”). Several years later, this morphological description was adopted and refined in the first French–American–British classification proposed in 1976, assigning this entity the M3 subtype among the eight recognized subtypes of AML [8].
3. THE ERA OF CHEMOTHERAPY AND GENETIC CHARACTERIZATION OF APL
3.1. Advances in Chemotherapy and Supportive Therapy
The introduction of chemotherapy for APL was the first turning point that dramatically changed the natural course of the disease. Since the landmark article by Bernard et al. [5], when they reported the first successful results with high-dose daunorubicin, chemotherapy for this leukemia has essentially been based on two main approaches: (1) AML-like chemotherapy, generally based on the combination of an anthracycline and cytarabine; and (2) Tailored therapy for APL, generally based on monotherapy with high-dose anthracycline. The latter was the strategy adopted by most groups in Europe. Most of the studies in the literature reported a high rate of complete remission (CR), ranging from 47% to 88% (Table 1). They also showed a higher rate of fatal bleeding and a lower rate of primary resistance compared to other subtypes of AML. The latter indicates a peculiar sensitivity of APL blasts to anthracyclines, which, in turn, is most likely related to absent or low expression of multidrug resistance mediated by the p170 glycoprotein and other minimal residual disease (MRD)-associated molecules [9–11]. Taken together, the results obtained by combining an anthracycline to cytarabine did not appear to improve CR rates (Table 1). It should be noted, however, that the only available randomized study comparing idarubicin alone with idarubicin plus cytarabine, carried out by the Gruppo Italiano Malattie Ematologiche dell’ Adulto (GIMEMA) group in the pre-ATRA era, showed better results with idarubicin alone in terms of event-free survival, although no difference was observed in the CR rate [12].
| References | No. of patients | CR (%) |
|---|---|---|
| Daunorubicin alone | ||
| Bernard et al. [5] | 34 | 55 |
| Marty et al. [18] | 83 | 64 |
| Petti et al. [118] | 30 | 67 |
| Sanz et al. [119] | 34 | 68 |
| Rodeghiero et al. [120] | 126 | 67 |
| Fenaux et al. [121] | 35 | 88 |
| Idarubicin alone | ||
| Avvisati et al. [122] | 27 | 81 |
| Avvisati et al. [12] | 131 | 76 |
| Anthracicline + Ara-C ± other | ||
| Ruggero et al. [123] | 13 | 54 |
| Daly et al. [124] | 15 | 73 |
| Arlin et al. [125] | 16 | 75 |
| Cordonnier et al. [126] | 57 | 53 |
| Kantarjian et al. [19] | 60 | 53 |
| Goldberg et al. [127] | 27 | 74 |
| Hoyle et al. [128] | 115 | 60 |
| Cunningham et al. [129] | 57 | 72 |
| Rodeghiero et al. [120] | 142 | 58 |
| Thomas et al. [130] | 67 | 49 |
| Fenaux et al. [131] | 21 | 86 |
| 18 | 66 | |
| Head et al. [132] | 45 | 71 |
| 96 | 47 | |
| Willemze et al. [133] | 80 | 54 |
Main reported series of APL treated with chemotherapy alone
For post-remission therapy during the pre-ATRA era, a variety of approaches were used, usually mimicking the strategy used for other AML subtypes. Thus, initial studies included consolidation courses of moderate intensity (re-inductions) with or without maintenance treatment. This approach was progressively changed with the introduction of higher intensity regimens, including autologous and allogeneic stem cell transplantation (SCT), as for the other AML subtypes. An European survey of SCT in APL analyzed the outcome of patients reported to the European society for Blood and Marrow Transplantation registry between November 1979 and December 1992 (mainly the period before ATRA in Europe) [13]. That study showed that both autologous and allogeneic transplantation were performed primarily in CR1 (69% and 81%, respectively) and roughly 45% of APL patients were cured by SCT. Although this treatment approach was still used for some years after the introduction of ATRA, the outstanding results obtained with the combination of ATRA and chemotherapy soon discouraged the use of SCT in first-line treatment. In fact, SCT has virtually ceased to play any role in CR1 and has been relegated to consolidation for patients in CR2 or beyond after salvage therapy for relapsed APL [14–17].
During that period, it is also interesting to mention two studies that suggested a survival advantage in patients with APL when they were administered maintenance treatment with 6-mercaptopurine and methotrexate [18,19].
Significant advances in supportive therapy over that period, particularly in the management of infections and transfusion support, resulted in increased CR rates by decreasing mortality from infection or bleeding during induction. In particular, massive transfusion of platelets and fresh plasma have been the cornerstone that has supported the prevention and treatment of bleeding diathesis in APL. Other measures, such as the use of heparin, tranexamic acid or other anticoagulant or antifibrinolytic treatments have not shown benefit to decrease the bleeding risk associated with APL coagulopathy and, therefore, are not currently recommended [14,15].
3.2. First Steps in Understanding the Biology and Genetics of APL
During the pre-ATRA era, there were several major breakthroughs on the biology and genetics of APL, such as the discovery of the t(15;17) translocation, the identification of the promyelocytic leukemia retinoic acid receptor alpha (PML-RARA) fusion gene resulting from this cytogenetic alteration, and the development of in vitro studies demonstrating the possibility of reversing malignancy by inducing differentiation of myeloid leukemia.
A chromosome abnormality in APL was first reported by the University of Chicago group led by Rowley, which was primarily interpreted as a partial deletion of the long arm of chromosome 17 [20]. A year later, that group discovered that it was actually a reciprocal translocation between the long arms of chromosomes 15 and 17 [21,22]. After a period of controversy about the specificity of this chromosome alteration, and the possibility of geographic differences in the incidence of this alteration in APL patients [23,24], the University of Chicago’s group definitively established that the t(15;17) with breakpoints at 15q22 and 17q21.1 may be found in every APL patient if optimal chromosome analysis is performed [25]. Today we know that, apart from the methodological flaws in cytogenetic studies, which were very common at that time, some patients have cryptic translocations with insertions or other more complex chromosomal exchanges that make detection of the t(15;17) difficult [26,27].
The discovery of the specific t(15;17) chromosomal translocation in APL contributed to the recognition of a less differentiated variant called microgranular, or M3 variant, a few years later [28]. This morphological variant, which accounts for approximately one third of APL cases, typically presents with leukocytosis with numerous circulating abnormal promyelocytes, but has the same cytogenetic and molecular features as the classical hypergranular APL (M3).
At the very beginning of the 90s, just in the transition period to the ATRA era, another crucial advance took place in the genetic characterization of APL, the discovery and cloning of the PML-RARA rearrangement, whose historical significance is unquestionable. The precedent for this discovery was the identification of the RARA gene at the 17q21 breakpoint implicated in the t(15;17) [29]. Cloning and sequencing of the RARA gene and its breakpoint is attributed to three research teams led, respectively, by de Thé et al. [30], Solomon et al. [31] and Pelicci [32], who published their studies almost simultaneously in the early 1990s. A year later, the PML gene (originally named MYL), located on chromosome 15q22, was completely sequenced by de Thé et al. [33] and Kakizuka et al. [34], who also simultaneously published their discovery in the same issue of Cell. It was later shown that this gene participates in the control of cell proliferation, apoptosis and senescence [35]. The cloning of the t(15;17) also enabled the design of polymerase chain reaction (PCR)-based strategies not only for a rapid diagnosis, but also for sensitive detection of MRD [36–40].
The circumstances that led to the use of ATRA as a treatment for APL have been extensively described by some of the protagonists of this story [41,42]. Certainly, some pioneering studies had suggested the in vitro differentiation capacity of teratocarcinoma cells [43] and, 10 years later, of neuroblastoma [44], as well as of a murine virus-induced erythroleukemia cell line when stimulated with dimethyl sulfoxide [45]. However, the greatest inspiring advance of differentiation therapy in leukemias was made by Sachs [46], who discovered that leukemic cells could be induced to differentiate under the action of certain agents, contradicting the dogma of the irreversibility of the leukemic cell. In the following years, a wide variety of agents, including retinoic acid, a derivative of vitamin A, demonstrated their ability to induce morphological and functional maturation of myeloid cells of the HL-60 line [47,48]. This cell line has many features in common with APL cells, but it does not carry the t(15;17) and only has one chromosome 17. Likewise, leukemic cells from patients with APL (M3) differentiate in vitro in the presence of retinoids [47,48]. These laboratory investigations did not take long to lead to anecdotal observations of patients with APL who showed clinical improvement or even CR, along with promyelocyte maturation (differentiation), when treated with 13-cis retinoic acid, a retinoic acid isomer other than ATRA [49–51].
In 1985, during an informal meeting of researchers from Shanghai with Laurent Degos of the Institute of Hematology at Saint Louis Hospital in Paris, the feasibility of treating leukemia by inducing differentiation with ATRA and/or low-dose cytosine arabinoside was raised and discussed. A few months later on that same year, given the availability of ATRA, just approved by the Shanghai Municipality for the treatment of skin diseases such as psoriasis and acne, the Shanghai group decided to try it in a 5-year-old girl with refractory APL [41]. Three weeks later, the girl achieved CR and received post-remission treatment with alternating ATRA and chemotherapy for one year. Since then, she has been in continuous CR. The success of this case encouraged further testing of the drug in other cases of APL, which marked the beginning of the ATRA era and represented the first model of targeted therapy for cancer.
4. THE ERA OF ATRA – OPTIMIZATION OF THE COMBINATION WITH CHEMOTHERAPY
The extension of the use of ATRA alone to six cases of APL (four newly diagnosed and two refractory to chemotherapy), reaching CR in all of them, was reported in 1987 in a Chinese journal [52]. However, it was not until a year later that the impressive results obtained by the Shanghai Institute of Hematology in 24 patients with APL (16 newly diagnosed and eight refractory cases) treated with ATRA alone were released to the rest of the world [41]. All patients achieved CR, but one required the addition of low-dose cytosine arabinoside to ATRA. From a privileged position, as one of the protagonists in the history, Degos [42] describes in his article “the history of APL” how they faced the challenge of treating the first APL patients with ATRA in France.
4.1. Early Results with ATRA Alone without Further Chemotherapy
After the seminal report by the Shanghai group [53], a few additional studies confirmed the efficacy of ATRA alone to achieve CR in patients with APL (Table 2). These confirmatory studies were carried out by a select group of institutions in the world, the Shanghai Institute of Hematology [54], the Jiangsu Institute of Hematology [55], the Hospital Saint-Louis in Paris [56,57], and the Memorial Sloan Kettering Cancer Center in New York [58]. Apart from the high rates of CR, all these studies described some differential features of induction with ATRA compared to chemotherapy. In contrast to chemotherapy, the administration of ATRA did not produce aplasia or alopecia, with few infections and a rapid improvement in coagulopathy. The gradual terminal differentiation of malignant promyelocytes in the bone marrow was also a very striking cytomorphological feature. However, it was soon found that patients who achieved CR generally relapsed within a few months, despite continuous ATRA treatment (median 5 months). This observation inspired the need to implement modifications in the therapeutic strategy.
| References | ATRA dose (mg/m2) | No. of patients | CR (%) | Relapse |
|---|---|---|---|---|
| Huang et al. [53] | 45–100 | 24* | 100 | 8 |
| Degos et al. [56] | 45 | 20 | 95 | 9 |
| Castaigne et al. [57] | 45 | 22 | 64 | 9 |
| Wang et al. [54] | 45–100 | 57 | 84 | NA |
| Warrell et al. [58] | 45 | 11 | 82 | 1 |
| Chen et al. [55] | 60–80 | 50 | 94 | 19 |
*Low-dose Ara-C was added to one patient.
NA, not available.
Induction treatment with ATRA alone for APL
4.2. ATRA Alone Followed by Chemotherapy
Following the initial experience with ATRA alone, a randomized [59] and several non-randomized studies [60–63] conducted in the early 1990s showed that patients receiving ATRA followed by anthracycline-based chemotherapy (with or without cytarabine) did significantly better, in terms of either CR or relapse rates, than those treated with ATRA alone or chemotherapy alone. The randomized study conducted by the European APL group (APL91 trial) compared chemotherapy alone (daunorubicin plus cytarabine) with ATRA followed by chemotherapy [59]. Although differences in the CR rate were not significant, the study was terminated early because of a significantly lower relapse rate and superior disease-free survival in the ATRA followed by chemotherapy arm. Those results were confirmed in successive analyses with a longer follow-up [64,65]. Subsequently, other randomized [66], but also non-randomized studies [67,68] in which chemotherapy was given after ATRA induction confirmed the advantage of this strategy compared to either ATRA or chemotherapy alone (Table 3).
| References | No. of patients | Anthracycline | CR (%) | OS (%) |
|---|---|---|---|---|
| Fenaux et al. [59] | 54 | DNR | 95 | 76 (4 years) |
| Frankel et al. [67] | 34 | IDA | 86 | 70 (3 years) |
| Tallman et al. [66] | 172 | DNR | 72 | 71 (3 years) |
| Asou et al. [68] | 62 | DNR, ACR, MTZ | 88 | 74 (4 years) |
| Fenaux et al. [69] | 109 | DNR | 95 | 81 (2 years) |
ATRA 45 mg/m2/day.
ACR, aclarubicin; DNR, daunorubicin; IDA, idarubicin; MTZ, mitoxantrone; OS, overall survival.
Studies with ATRA alone for induction followed by chemotherapy
4.3. ATRA plus Chemotherapy
The simultaneous combination of ATRA and chemotherapy was shown to be superior to their sequential administration (ATRA followed by chemotherapy) in a randomized study of the European APL group [69]. The advantage of this approach was further confirmed in other large, uncontrolled, multicenter trials carried out during the second half of the 90s [68,70–75] (Table 4). Based on these studies, the European LeukemiaNet established the combination of ATRA plus anthracycline-based chemotherapy for induction, followed by 2–3 courses of anthracycline-based chemotherapy for consolidation, as the standard of care for newly diagnosed APL [14].
| References | ATRA (mg/m2/day) | Induction chemotherapy (mg/m2/day) | Consolidation courses |
|---|---|---|---|
| Mandelli et al. [70] | 45 until CR | IDA 12 × 4 days | 3 |
| Estey et al. [71] | 45 until CR | IDA 12 × 4 days | 2 |
| Fenaux et al. [69] | 45 until CR | DNR 60 × 3 days | 2 |
| Ara-C 200 × 7 days | |||
| Sanz et al. [73] | 45 until CR | IDA 12 × 4 days | 3 |
| Burnett et al. [72] | 45 until CR | DNR 60 × 3 days | 3 |
| DAT versus ADE |
DAT, daunorubicin, Ara-C, and thioguanine; DAE, Ara-C, daunorubicin, and etoposide.
Studies with ATRA plus chemotherapy for induction and consolidation
4.4. Definition of Relapse-Risk Categories and Adoption of Risk-Adapted Strategies
The results obtained with the simultaneous administration of ATRA and chemotherapy provided an important source for the investigation of prognostic factors to be used for treatment stratification. In this regard, the definition of relapse risk in a joint study of the GIMEMA and Programa Español de Tratamientos en Hematología (PETHEMA) groups was critical for the design of risk-adapted strategies [70,71,73,74]. In this way, a gradual intensity could be given to intensify treatment only in patients at higher risk of relapse, while a reduction in the dose-intensity of chemotherapy in patients with lower risk could reduce toxicity without compromising cure rates. The results of both GIMEMA and PETHEMA studies using such approach showed significant improvement of outcome [75–78]. Since then, there is a tendency to design risk-adapted strategies to modulate treatment intensity in consolidation according not only to age, assuming that older patients are more vulnerable to toxicity, with higher rates of neutropenic sepsis and increased treatment-related mortality, but also to predefined risk factors for relapse, particularly presenting high WBC counts [15,16].
Another interesting strategy for tailoring post-remission treatment that has been proposed is the use of a reliable sequential real-time quantitative PCR (RQ-PCR) monitoring of PML-RARA [79]. Encouraging results have been reported with such strategy when coupled with pre-emptive therapy (MRD-driven therapy), but it remains to be established whether this approach can be considered today as a standardized, cost-effective, and universally available method for routine laboratories.
4.5. Understanding the Molecular Mechanisms involved in APL Leukemogenesis and Response to Targeted Therapy
After the genetic studies reported in the early 1990s, the availability of cell lines and transgenic mouse models harboring the PML-RARA fusion gene promoted research on APL leukemogenesis, and unraveled the molecular mechanisms involved in the transcriptional repression induced by the PML-RARA protein, as well as the response to ATRA and ATO [80–83]. Several subsequent studies have led us to know that the oncogenic activity of PML-RARA is exerted through both dominant-negative and gain-of-function effects [84,85]. The PML-RARA fusion protein maintains the ability to multimerize with various classes of proteins, enhancing their binding to corepressors and acting as a super-repressor insensitive to physiological concentrations of ATRA [83,86]. Thus, PML-RARA represses the transcription of several genes involved in myeloid differentiation through the altered recruitment of histone deacetylases [87]. In addition, PML-RARA–RXR complexes mainly recognize atypical RARA and interact with genomic regions, which confers a proliferative and survival advantage to leukemic cells [88,89].
A few years later, the mechanisms involved in the response to targeted therapy with ATRA, but also with ATO, which are capable of restoring PML-RARA functions, have also been unraveled. Thus, ATRA induces proteasome-mediated PML-RARA degradation and transcriptional activation as a consequence of the release of various corepressors and interaction with a series of coactivators [90,91], while ATO induces post-transcriptional modifications in the second B box (B2) domain of the PML moiety, which results in the restoration of nuclear bodies (NB) and PML-RARA degradation [92,93].
4.6. Advances in the Management and Supportive Therapy
In addition to the improvements that occurred in the management of patients with acute leukemia in general, mainly in transfusion support as well as in the prevention and treatment of infections, some others specifically benefited patients with APL, such as the recognition of the differentiation syndrome and its easy reversibility when it is treated early and appropriately, and a better understanding of the ATRA catabolic process, among others.
Although cases in which an increase in leukocytes was accompanied by fever and dyspnea, and occasionally kidney failure, had already been reported among the first patients treated with ATRA [57], it was in 1992 when the differentiation syndrome (DS), originally described as retinoic acid syndrome, was first defined [53]. The full-blown syndrome is characterized by unexplained fever, weight gain, peripheral edema, dyspnea with interstitial pulmonary infiltrates, pleuropericardial effusion, hypotension, and acute renal failure. Today we know that, in the context of treatment with ATRA plus chemotherapy, approximately a quarter of patients present some symptom or sign of this syndrome [94], whereas the incidence may be somewhat higher with the combination of ATRA plus ATO. Since the New York group reported an excellent response to corticosteroids [53], the early administration of dexamethasone has become the cornerstone for the management of DS and decrease the DS-associated mortality to 1% [94].
Another very interesting aspect that has contributed to optimizing treatment with ATRA was the early observation that patients treated only with this agent achieved short-term remissions, but that after a therapeutic pause it could occasionally be observed that resistance was reversible [62]. The most widely accepted hypothesis is that this resistance was probably due, in almost all patients, to a catabolic process that reduces the concentration of ATRA in the body. In fact, pharmacological studies were able to demonstrate a substantial decrease in ATRA plasma levels after only a few days of treatment, remaining at low levels even if the dose was significantly increased [95]. Moreover, it was shown that ATRA induces an increase in the cytochrome P450 activity which, in turn, is also involved in the catabolism of ATRA. These findings led to combine ATRA with chemotherapy and use ATRA intermittently rather than continuously in maintenance therapy.
Historically, the advances in the management of APL coagulopathy must also be highlighted. Physicians soon realized that chemotherapy exacerbated bleeding diathesis and signs of coagulopathy and, therefore, the risk of early death. Whether the coagulopathy of APL is due to a disseminated intravascular syndrome with reactive fibrinolysis, to a primary fibrinolysis, or both pathogenetic mechanisms simultaneously, is still a matter of controversy. Beyond the pathogenesis of APL-associated coagulopathy, massive transfusion of platelets and fresh plasma have been the most effective tool for the prevention and treatment of bleeding diathesis in APL.
In his excellent historical review, Degos describes how the spectacular advances produced from the mid-1980s to the early 1990s, both in the field of therapy and in the understanding of many biological aspects of APL, led Mandelli to organize a first international meeting in Rome in 1993, under the title “APL - A curable disease?”. Since then, every 4 years, this meeting in Rome has become the most important international scientific summit to review the continuous advances in this disease.
5. THE ERA OF ATO
5.1. Early Results with ATO Alone
The introduction of arsenic derivatives in the treatment of APL, as occurred with ATRA, was made in China. In the early 1970s, a group from Harbin Medical University in Manchuria identified ATO as a potential anticancer drug, and began to use an arsenic compound to treat a variety of malignancies. But it was not until 1992, that this group reported that 21 out of 32 patients with APL entered CR with an impressive 30% survival rate after 10 years with Ailing-1 (anticancer-1 or Al-1), an anticancer solution containing 1% ATO [96]. These results were subsequently confirmed by the Shanghai group [97–99].
5.2. Confirmation of ATO as Standard Treatment for Relapsed APL
The impressive results from pioneering studies carried out in China encouraged the design of a pilot study conducted at Memorial Sloan Kettering Cancer Center in New York, which confirmed the effectiveness of ATO in inducing CR in 11 of 12 patients with relapsed APL after extensive prior therapy [100]. These results prompted a multicenter trial in the USA in which ATO was administered as salvage therapy in relapsed patients after ATRA and anthracycline-based chemotherapy [101]. Thirty-four of 40 patients (85%) achieved CR with ATO alone and 86% achieved molecular CR after consolidation therapy. These results established ATO as a highly effective therapy for relapsed APL patients and led to the approval of this drug by the FDA in September 2000 for the treatment of relapse/refractory APL.
5.3. The First Pilot Studies with ATO-based Regimens for Newly Diagnosed APL
Encouraged by the results with ATO in relapsed patients, the Harbin and Shanghai groups expanded the use of this agent to newly diagnosed patients with APL with promising results [99,102]. On the basis of this experience, a few pilot studies in newly diagnosed APL patients treated with ATO alone or in combination with ATRA were carried out in China [103], Iran [104], India [105], and the USA [106]. The CR rate in these studies ranged from 86% to 95%. However, it should be noted that ATO was combined with ATRA [103,106] and/or chemotherapy [103,105] and/or gemtuzumab ozogamicin [106] in a variable proportion of patients, particularly those presenting with hyperleukocytosis (Table 5).
| References | No. of patients | Induction therapy | CR (%) | DFS (%) |
|---|---|---|---|---|
| Zhang et al. [102] | 30 | ATO | 73 | NA |
| Niu et al. [99] | 11 | ATO ± CHT | 73 | 100 (1 year) |
| Shen et al. [103] | 41 | ATO ± ATRA | 90–95 | 87–100 (2 years) |
| Ghavamzadeh et al. [104] | 94 | ATO | 86 | 64 (2 years) |
| Mathews et al. [105] | 72 | ATO | 86 | 87 (3 years) |
| Estey et al. [106] | 44 | ATO + ATRA ± CHT | 89 | 84 (2 years)* |
Relapse-free survival.
CHT, chemotherapy; DFS, disease-free survival.
First pilot studies with ATO-based regimens for newly diagnosed APL patients
5.4. Confirmation of ATO-based Approach as Standard Treatment for Newly Diagnosed APL
Despite the high efficacy reported in the studies mentioned above, the 2009 European LeukemiaNet recommendations highlighted the absence of properly designed comparative studies of ATO-based versus the standard ATRA plus chemotherapy approach in terms of efficacy, safety, and cost-effectiveness [69]. Therefore, the recommendation was that, in the meantime, the use of ATO-based regimens should be restricted to patients included in clinical trials or for those in whom chemotherapy (especially anthracyclines) was contraindicated. It was also recognized that, in countries where locally produced arsenic compounds provide a more affordable treatment approach than ATRA plus chemotherapy, a cheaper ATO, produced under good quality control, would be effective in curing many patients with APL.
Two multicenter phase 3 trials were set up to compare the efficacy and safety of ATRA plus ATO versus ATRA plus chemotherapy [107,108]. One trial was conducted by the Italian cooperative GIMEMA in collaboration with the German-Austrian AML Study Group and Study Alliance Leukemia cooperative group, and compared ATRA plus ATO versus ATRA plus chemotherapy (AIDA regimen) in patients with low-to-intermediate risk APL (WBC count ≤10 × 109/L) [107]. The other randomized trial conducted by the National Cancer Research Institute cooperative group included all relapse risk categories [108]. Based primarily on the results of these two randomized trials and the long-term results of a nonrandomized study conducted at the MD Anderson Cancer Center in Houston [109], the combination of ATRA plus ATO has been adopted worldwide as the new standard of care for patients with non-high-risk APL (WBC count ≤10 × 109/L) [15,16,110,111].
6. CURRENT PERSPECTIVES AND FUTURE DIRECTION
Once the extraordinary advances toward the cure of APL have been consolidated, two important issues remain to be resolved in a small fraction of patients with APL: early death and relapse. While these great challenges are solved, some other advances, whose development is quite predictable, could be incorporated into clinical practice in the short or medium term. Among them is the optimization of treatment in patients at high risk of relapse, seeking the most appropriate combination of the three currently available agents, ATRA, ATO, and chemotherapy. The use of oral arsenic derivatives instead of intravenous ATO is already a reality in China, and we can envisage that sooner or later it will also be the case in the rest of the world. In fact, several oral formulations of arsenic derivatives are currently in different stages of clinical development.
In China, four types of oral arsenic formulations have been used in APL: tetra-arsenic tetrasulfide (As4S4), ATO (As2O3), Qinghuang powder (realgar and indigo naturalis), and RIF (realgar, indigo naturalis formula), radix salviae miltiorrhizae, and radix pseudostellariae) [112]. Among them, only RIF is commercially available in China, but it has not been licensed elsewhere. This compound has not only demonstrated clinical efficacy comparable to intravenous ATO in two randomized studies in newly diagnosed APL, but also encompassed a better safety profile, improved quality of life, and lower costs [113,114]. In addition, at least three different formulations of oral ATO are being explored. A single-center study from Hong Kong using an oral formulation of ATO has provided excellent long-term outcomes in relapsed APL [115]. Data from a comparative bioavailability study developed in Australia (APML5; ACTRN12616001022459) have shown promising evidence of average bioequivalence between encapsulated oral and intravenous ATO [116]. Finally, a multicenter phase 1 study conducted in the USA with another oral ATO formulation (clinical trial registration # NCT03048344) has demonstrated that ORH-2014 at 15 mg is safe, bioavailable, and provides the required arsenic exposure compared to intravenous ATO at the approved dose (0.15 mg/kg) [117]. Certainly, these oral ATO formulations should be tested in prospective randomized studies. If their efficacy and safety profile is comparable to that of the intravenous preparations, oral ATO may be explored in an outpatient approach for the treatment of APL, especially for post-remission treatment.
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
AUTHORS’ CONTRIBUTION
Both authors contributed equally.
REFERENCES
Cite this article
TY - JOUR AU - Miguel A. Sanz AU - Eva Barragán PY - 2021 DA - 2021/07/19 TI - History of Acute Promyelocytic Leukemia JO - Clinical Hematology International SP - 142 EP - 152 VL - 3 IS - 4 SN - 2590-0048 UR - https://doi.org/10.2991/chi.k.210703.001 DO - 10.2991/chi.k.210703.001 ID - Sanz2021 ER -