
Virtual Screening and Inhibition of Middle East Respiratory Syndrome Coronavirus Replication by Targeting Papain-like Protease
 , Byoung Kwon Park3, , Mohamed A. Morsy4, 5, , Katharigatta N. Venugopala4, 6, , Kentaro Oh-hashi7, Mohammed Al-Nazawi1, Hyung-Joo Kwon3,
, Byoung Kwon Park3, , Mohamed A. Morsy4, 5, , Katharigatta N. Venugopala4, 6, , Kentaro Oh-hashi7, Mohammed Al-Nazawi1, Hyung-Joo Kwon3, - DOI
- 10.2991/dsahmj.k.210921.001How to use a DOI?
- Keywords
- MERS-CoV; papain-like protease; antiviral agents; virtual screening; plaque
- Abstract
Infection by the emerging, potentially zoonotic Middle East Respiratory Syndrome Coronavirus (MERS-CoV) presents a severe health hazard to humans and is often fatal. Given the lack of particular medicines against MERS-CoV, drug discovery studies are needed to bridge this knowledge gap. In this study, we introduce virtual screening-guided identification of MERS-CoV Papain-like Protease (PLpro)-binding drugs. After a two-step virtual screening method, enzyme assays and antiviral testing with a MERS-CoV plaque reduction assay were used to further investigate the five compounds with the highest computational score. The top five screened compounds showed a 10.2–40% decrease in MERS-CoV PLpro activity. The top two compounds showed promising inhibition of MERS-CoV replication, reducing virus plaque formation by 30.6% and 24%. Compounds 1 and 4 in this study can be further modified to target binding with MERS-CoV PLpro active triad residues. Furthermore, the compounds produced stable interaction with the protein and protein conformation. With their reported inhibition of MERS-CoV enzyme and virus replication, supported by favorable absorption, distribution, metabolism, and excretion and toxicity profiles, the two reported benzimidazole and piperazine derivatives could be considered lead compounds against MERS-CoV.
- Copyright
- © 2021 Dr. Sulaiman Al Habib Medical Group. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
Middle East Respiratory Syndrome Coronavirus (MERS-CoV) is an emerging viral infection responsible for severe respiratory and systemic illness [1]. MERS-CoV is propagated by both zoonotic and human-to-human transmission mechanisms [2,3]. CoVs were once thought to be a component of the common cold’s causative agents. However, in recent decades, more serious strains such as the Severe Acute Respiratory Syndrome CoV (SARS-CoV), MERS-CoV, and SARS-CoV-2 have emerged [4].
The CoV polyprotein encodes approximately 14–16 Nonstructural (NS) proteins. The Main Protease (Mpro) and Papain-like Protease (PLpro), two virally encoded proteases, digest the viral polyprotein and aid in the release of virally encoded enzymes and proteins required for the completion of CoVs replication cycle [5]. PLpro was identified as a valid target for identifying drugs against MERS-CoV [6] and SARS-CoV [7]. Of particular interest, inhibition of PLpro is believed to affect virus replication because of its multiple protease, deubiquitination and de-ISGylation activities [8,9]. The structural components of deubiquitination and de-ISGylation include a hand-shaped central domain and ubiquitin-like domain [10–12].
Several research groups have focused on MERS-CoV PLpro to discover novel inhibitors. [13], and several previous trials were performed to identify new small molecule inhibitors of MERS-CoV [6,14–17]. Disulfiram [18], thiopurine, and mycophenolic acid [19] were recommended for MERS-CoV inhibition through interference with MERS-CoV PLpro activity. Our group is working to discover new anti-CoV compounds. Small molecule inhibitors were discovered for the virus entry process [20,21], and fusion peptide inhibitors were identified for MERS-CoV and SARS-CoV-2 [21,22].
Because of the significance of SARS-CoV-2 PLpro as a drug target, several studies have been conducted to rapidly repurpose approved drugs for use in biological and inhibitory aspects of SARS-CoV-2 PLpro [23,24]. In addition, various chemical compounds and their derivatives were extensively investigated, for example, naphthyl derivatives [25], flavonol derivatives [26], thiopurine [27], aminopurines [6], tropane alkaloids [28], and various FDA-approved drugs [29,30].
A small-molecule inhibitor was first identified in our previous work to discover new anti-MERS-CoV PLpro drugs [6]. The aim of this study includes (1) extension of the previous work and introduction of new inhibitors and (2) investigation of the effect of compounds on MERS-CoV replication in cell cultures.
2. MATERIALS AND METHODS
2.1. Compound Library and Molecular Modeling
2.1.1. Ligand preparation
Commercially available focused screening libraries from ChemBridge Inc. (San Diego, CA, USA) were used to generate a chemical search space. The library contains the following sets: specific lipophilic molecules of Central Nervous System (CNS) targets, CombiSet, diverse set, ion channel, kinase-specific compound, antimicrobial set, epigenetic, mitogen-activated protein kinase, and anticancer. The compound sets included general and targeted libraries, lead-like, drug-like, lipophilic, hydrophilic, and varied chemical scaffolds. Among the focused libraries, the CNS library was used, as it demonstrates high penetrability of body barriers such as the blood–brain barrier. Furthermore, the antimicrobial library comprised a set of microbe-specific compounds, including antivirals. All compounds were desalted and 3D optimized by the standard settings of LigPrep software. A summary of experimental procedures is provided in Figure 1A.

The flow, stages and screening studies against MERS-CoV PLpro. (A) Flow chart showing the stages of investigations, comprising compounds library preparations, two phases of docking and multiple assays. (B) (B-A) The composition of MERS-CoV PLpro. (B-B) Apo MERS-CoV PLpro, MERS-CoV PLpro showing the sites of docked compounds after the virtual screening. (B-C) Surface representation of MERS-CoV PLpro showing the sites of docked compounds after virtual screening. (B-D) Focused view showing the catalytic site of PLpro occupied with the docked compounds.
2.1.2. Drug-likeness and ligand-based absorption, distribution, metabolism, and excretion/toxicity prediction
Absorption, Distribution, Metabolism, and Excretion (ADME) are pharmacokinetic descriptors for new compounds. ADME highlights the drug levels and drug exposure kinetics in tissues, as well as the compound’s efficiency and pharmacological activity. QikProp version 4.2 (Schrödinger LLC Maestro package, New York, NY, USA) was used to describe drug-likeness and predicted pharmacokinetics and toxicity. The selected molecular descriptors were as described previously [20]. The selected descriptors comprise MW, H-bond donor, H-bond acceptor, oral absorption % in humans, the number of violations of Lipinski’s rule of five, octanol/water partition coefficient (QPlog P o/w), Caco-2 cell permeability, and Madin–Darby Canine Kidney (MDCK) cell permeability.
2.1.3. Preparation of MERS-CoV PLpro structure
The preparation of protein structure was as previously described [6,20]. Briefly, the MERS-CoV PDB ID 4rf0 was used as a template for protein docking. The structure errors were initially fixed, and water was removed. The protein structure was optimized at physiological pH and energy minimized using an OPLS2005 force field. The docking grid was built by generating a grid box at 20 Å around the cocrystalized ligand.
2.1.4. Virtual screening
To acquire comprehensive insight into the putative inhibitor, a two-step docking procedure was used. The glide docking of Schrödinger LLC Maestro package (New York, NY, USA) was initially configured at the Standard Precision (SP docking). The results were ranked by docking score. Compounds with a docking score of –6.5 or higher were retrieved. These retrieved compounds were reanalyzed using an extra-precision docking protocol (XP docking), and the resulting top five compounds were further evaluated by biochemical enzyme activity, assays.
2.2. PLpro Inhibition Assay
Analyzing the effect of compounds on MERS-CoV PLpro activity was performed as previously described [6]. Enzyme activity was traced using the fluorogenic substrate Cbz-Arg-Leu-Arg-Gly-Gly-7-amino-4-methyl coumarin in the presence or absence of inhibitor compounds.
2.3. MERS-CoV Plaque Reduction Assay
2.3.1. Cell line and virus
The cell culture techniques conducted were as previously described [20,31]. African green monkey kidney cells (Vero cells) were maintained in an appropriate culture containing antibiotics. MERS-CoV (CoV/KOR/KNIH/002_05_2015) was obtained from the Korea Centers for Disease Control and Prevention (Permission No. 1-001-MER-IS-2015001).
2.3.2. Plaque reduction assay
The plaque reduction assay was performed as previously described [20,31]. In brief, MERS-CoV was mixed with each compound (at a final compound concentration of 10 µM) for 30 mintes at 37°C. The virus–drug mixture was applied to Vero cells and incubated for 30 min. The degree of plaque reduction seen following crystal violet staining was then used to determine the presence of active MERS-CoV. Compounds 1 and 4 were dissolved in Dimethylsulfoxide (DMSO) and diluted in Phosphate-buffered Saline (PBS) watery solution. The background effect of residual DMSO was also determined; then the number of plaques produced for each compound was compared to the normalized value after DMSO addition.
2.3.3. Molecular dynamics simulation
Molecular Dynamics (MD) simulation was performed as previously described with slight modifications [6,32]. The MD run was performed by the Desmond module of the Schrödinger LLC package. NPT simulation was adopted and frames were collected every 100 ps for analysis. The System Builder tool was used to prepare the system. The simulation was extended for 50 ns. The collected data comprised the Root Mean Square Deviation (RMSD) of the protein and ligands, the Root Mean Square Fluctuation (RMSF), the number of hydrogen bonds during the simulation, and the binding energy calculations. The Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) method was used to calculate the binding free energies of inhibitors with the PLpro.
3. RESULTS
3.1. Virtual Screening
Compounds with docking scores of −6.5 or higher were retrieved. A total of 433 compounds were shortlisted, and a summary of the statistics of the obtained compounds is provided in Table 1. The docking site selection was based on the original cocrystallized ligand position and conclusions drawn from prior PLpro catalytic site research (Figure 1B). The ranges for compound values were as follows: (1) clog P, −2.29 to 4.98; 2) hydrogen bond acceptor, 1–7; (3) hydrogen bond donor, 0–4; (4) docking score, −8.282 to −6.5; (5) ligand efficiency, −0.435 to −0.155; (6) lipophilic score, −3.479 to −0.839; (7) hydrogen bond score, −1.159 to 0.00; (8) van der Waals interactions, −49.22 to −14.32; and (9) electrostatic interactions, −21.81 to −2.64. The compounds were subjected to XP docking and the top five compounds were retrieved (Table 2). The compounds’ docking scores ranged from −8.2 to −8.4.
| clog P | Hacc | Hdon | Docking score | Glide lipo | Glide hbond | Glide evdw | Glide ecoul | Glide energy | Glide ligand efficiency | |
|---|---|---|---|---|---|---|---|---|---|---|
| Number of values | 433 | 433 | 433 | 433 | 433 | 433 | 433 | 433 | 433 | 433 |
| Minimum | −2.29 | 1 | 0 | −8.282 | −3.479 | −1.159 | −49.27 | −21.81 | −59 | −0.435 |
| 25% percentile | 1.693 | 3 | 1 | −6.858 | −2.654 | −0.693 | −39.3 | −12.78 | −48.5 | −0.28 |
| Median | 2.737 | 4 | 1 | −6.709 | −2.389 | −0.58 | −35.52 | −10.27 | −45.9 | −0.259 |
| 75% percentile | 3.651 | 5 | 2 | −6.585 | −2.049 | −0.434 | −31.34 | −7.932 | −42.85 | −0.24 |
| Maximum | 4.98 | 7 | 4 | −6.503 | −0.839 | 0 | −14.32 | −2.64 | −30.83 | −0.155 |
| Mean | 2.596 | 4.15 | 1.471 | −6.763 | −2.332 | −0.548 | −34.95 | –10.59 | −45.54 | −0.2621 |
| Std. deviation | 1.386 | 1.117 | 0.7327 | 0.2373 | 0.4506 | 0.2151 | 5.749 | 3.686 | 4.243 | 0.03325 |
| Std. error of mean | 0.066 | 0.0536 | 0.0352 | 0.0114 | 0.02165 | 0.0103 | 0.276 | 0.177 | 0.2039 | 0.001598 |
| Lower 95% CI of mean | 2.465 | 4.045 | 1.402 | −6.785 | −2.375 | −0.568 | −35.5 | −10.93 | −45.94 | −0.2652 |
| Upper 95% CI of mean | 2.727 | 4.256 | 1.54 | −6.74 | −2.29 | −0.527 | −34.41 | −10.24 | −45.14 | −0.2589 |
| Sum | 1,116 | 1,797 | 637 | −2.928 | −1,010 | −237. | −15.135 | −4.584 | −19,719 | −113.5 |
CI, confidence interval.
Descriptive statistics of the top compounds after virtual screening against the MERS-CoV PLpro PDB ID 4rf0. The table includes the compound ID, clog P, the number of hydrogen acceptor (Hacc), the number of hydrogen bond donor (Hdon), the docking score, the glide ligand efficiency, lipophilic interactions, hydrogen bonds score, glide van der Waals interactions, coulombic forces, and docking energy
| # | Structure | Mol. weight | Mol. formula | Molecular name | clog P | Hdon | Hacc | Docking score |
|---|---|---|---|---|---|---|---|---|
| 1 | 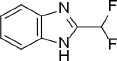 |
168.1 | C8H6F2N2 | 2-(Difluoromethyl)-1H-benzimidazole | 1.76 | 1 | 1 | −8.4645 |
| 2 | 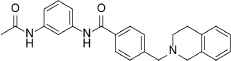 |
389.5 | C18H16FN3O2S2 | N-[2-({2-[(2-Fluorophenyl)amino]-2-oxoethyl}thio)-1,3-benzothiazol-6-yl]propanamide | 3.58 | 2 | 3 | −8.3508 |
| 3 | 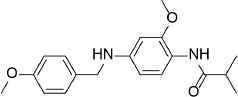 |
328.4 | C19H24N2O3 | N-{2-Methoxy-4-[(4-methoxybenzyl)amino]phenyl}-2-methylpropanamide | 2.425 | 2 | 3 | −8.3257 |
| 4 | 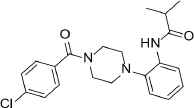 |
385.9 | C21H24ClN3O2 | N-{2-[4-(4-Chlorobenzoyl)-1-piperazinyl]phenyl}-2-methylpropanamide | 2.592 | 1 | 2 | −8.2947 |
| 5 | 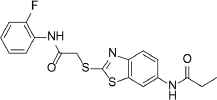 |
399.5 | C25H25N3O2 | N-[3-(Acetylamino)phenyl]-4-(3,4-dihydro-2(1H)-isoquinolinylmethyl)benzamide | 3.56 | 2 | 3 | −8.2195 |
Compounds with the highest docking score after virtual screening studies. The structure was drawn by ChemDraw software (CambridgeSoft, Cambridge, MA, USA). The molecular descriptors were obtained from the virtual screening compounds SD file data
3.2. Compound ADME and Toxicity Descriptors
The retrieved top compounds showed favorable pharmacokinetics and drug-likeness descriptors. There was no violation of Lipinski’s rule of five as observed by the optimal compounds’ molecular weight and hydrogen bond donor and acceptor numbers (Table 3). In addition, there was an estimated high oral absorption rate (93–100%) and improved cell penetrability.
| # | CNS | MWa | DonorHBb | AccptHBc | %Human oral absorptiond | Rule of Fivee | QPlog Po/wf | QPPCacog | QPPMDCKh |
|---|---|---|---|---|---|---|---|---|---|
| C1 | 1 | 168 | 1 | 1.5 | 100 | 0 | 2.396 | 3,699.071 | 6,286.69 |
| C2 | 1 | 399 | 2 | 7 | 93.3 | 0 | 3.941 | 263.012 | 129.209 |
| C3 | 0 | 328 | 2 | 5 | 100 | 0 | 4.24 | 3,976.174 | 2,199.384 |
| C4 | 0 | 385 | 1 | 6.5 | 100 | 0 | 4.517 | 2,514.447 | 3,308.723 |
| C5 | –1 | 389 | 2 | 6.5 | 100 | 0 | 3.836 | 714.611 | 1,310.845 |
Acceptable ranges are:
MW <500.
Hbond donor <5.
Hbond acceptor <10.
Oral absorption % in human, >80% is high.
The number of violations of Lipinski’s rule of five, maximum 3.
Octanol/water partition coefficient (QPlog Po/w) <5.
Caco-2 cell permeability >500 great.
MDCK cell permeability >500 is high.
MW, molecular weight.
Selected in silico pharmacokinetic and ADME descriptors for the top retrieved compounds after virtual screening
3.3. Enzyme Assay
The enzyme assay was based on the inhibition of cleavage of the florigenic substrate. All compounds at 40 µM inhibited MERS-CoV activity within a range of 10–40%. Compounds 1 and 4 showed the highest inhibition rate of 40% and 32%, respectively, whereas compounds 2, 3, and 5 showed 10.2%, 12%, and 14% activity reduction. To gain more insights into the potency of the compound, IC50 was measured by the estimation of enzyme kinetics data in the presence of different inhibitor concentrations. The estimated IC50 values were 22 ± 6.2, 88 ± 11.3, 80 ± 9.6, 12 ± 1.9, and 76 ± 15.6 µM for compounds 1–5, respectively. The potency of compounds was in the order 4 > 1 > 5 > 3 > 2. Based on these results, compounds 1 and 4 were examined in the MERS-CoV replication inhibition assay.
3.4. Plaque Reduction Assay
There were 484 normalized plaques in control wells. Compounds 1 and 4 reduced the normalized plaque counts to 363 and 368, respectively, indicating the reduced MERS-CoV replication by 30.6% and 24% (Figure 2 and Table 4).
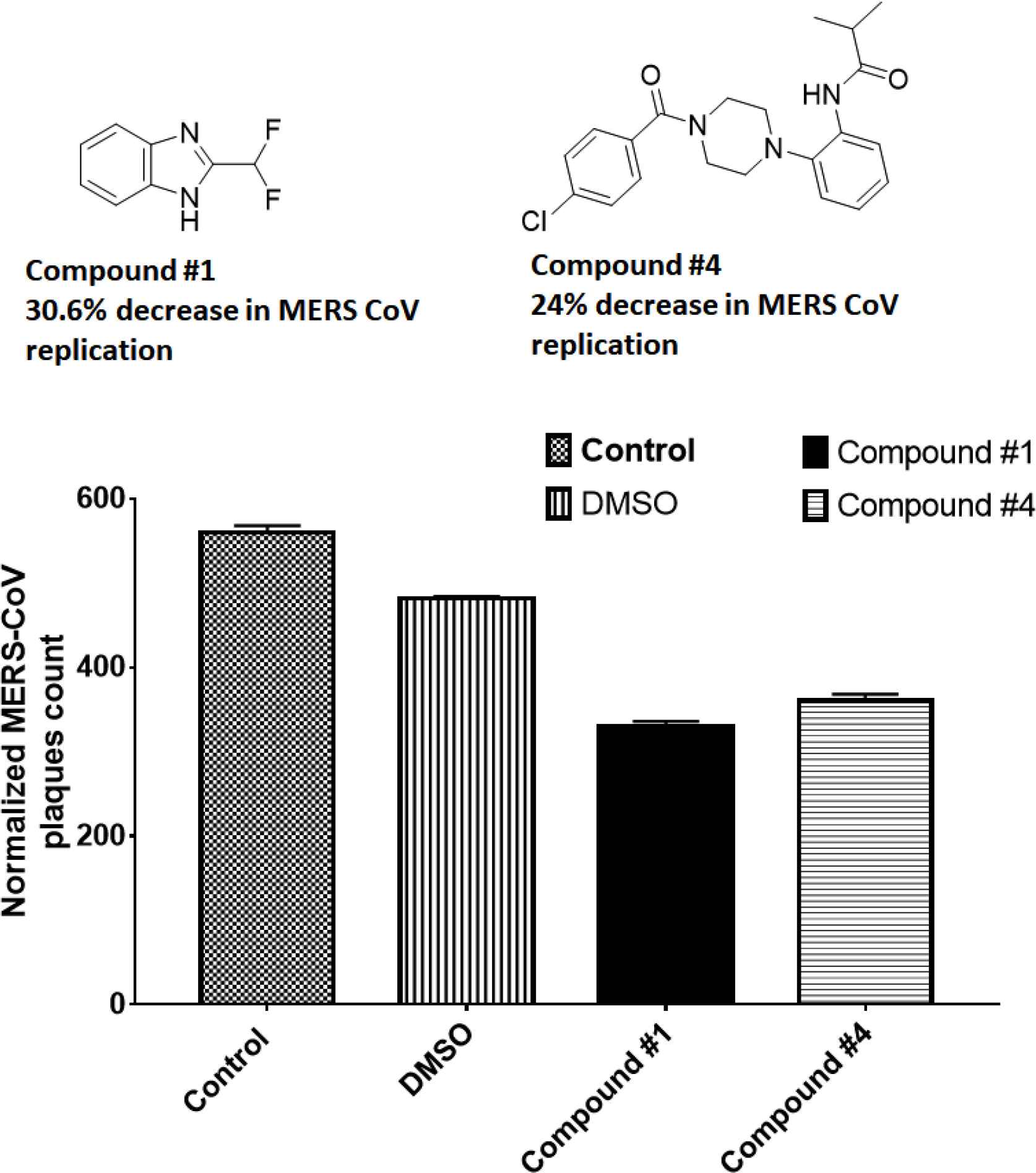
Screening of the inhibitors against MERS-CoV infection. The plaque reduction assay was performed with compounds 1 and 4. Prior to infection with MERS-CoV, the virus was incubated with each compound (10 µM) for 30 min at 37°C and then added to Vero cells. After incubation for 4 days in DMEM/F12 containing 0.6% oxoid agar, the plaques were revealed by crystal violet stain then counted. DMEM, Dulbecco’s modified Eagle’s medium.
| Total plaque no. | Normalized | % Plaque | |
|---|---|---|---|
| PBS | 142 | 568 | 117.3554 |
| 10% DMSO | 121 | 484 | 100 |
| C1 | 84 | 336 | 69.42149 |
| C4 | 92 | 368 | 76.03306 |
DMEM, Dulbecco’s modified Eagle’s medium.
Inhibition of MERS-CoV replication by the plaque assay. The plaque reduction assay was performed with compounds 1 and 4. Prior to infection with MERS-CoV, the virus was incubated with each compound (10 µM) for 1 h at 37°C and then added to Vero cells. After incubation for 4 days in DMEM/F12 containing 0.6% oxoid agar, the plaques were revealed by crystal violet stain then counted
3.5. MD Simulation
The RMSD of ligands and proteins was used to determine how far the atoms had strayed from the original structure (prior to simulation) (Figure 3). The protein RMSD is shown on the left Y-axis and represents the changes in atom location compared to the original structure throughout 50 ns of simulation. The RMSD changes in all compounds were almost all within the range of 1–2, acceptable values for globular proteins. The RMSD increased over the first few ns prior to becoming noticeably stable throughout the simulation (blue trace).
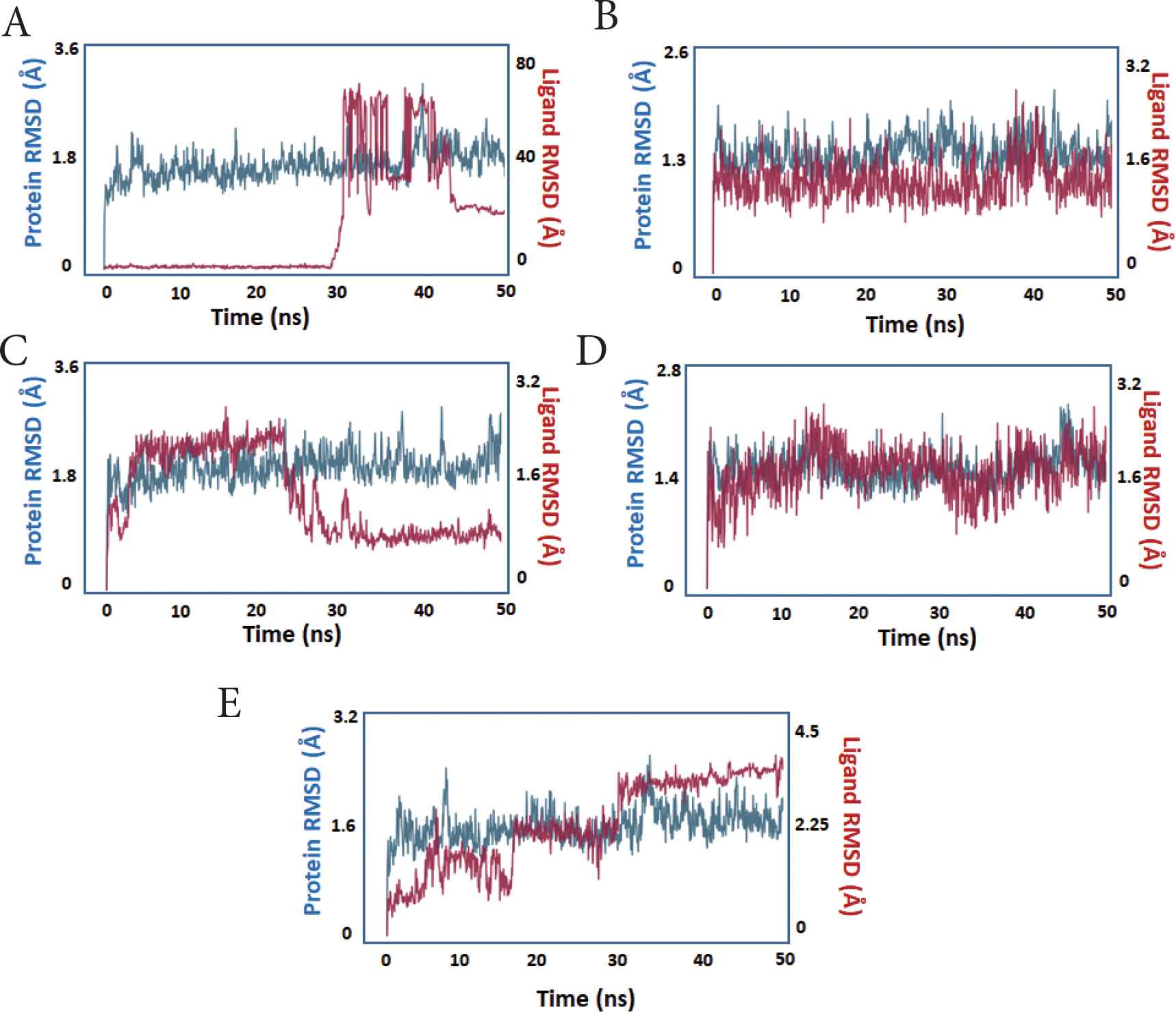
The protein and ligand RMSD during simulation. X-axis: time in nanoseconds. Y-axis: estimated RMSD in Angstroms. The protein trace is blue, whereas the ligand trace in red. (A) compound 1, (B) compound 2, (C) compound 3, (D) compound 4, (E) compound 5.
During the simulation, the ligand RMSD of the compounds was evaluated, which represents the state of ligands and their stability within the protein’s active site. With the exception of compound 5, all of the compounds showed measurable stability inside the PLpro active site, as seen by the ligands’ decreased RMSD relative to the protein (red trace).
To assess local changes in the protein in response to the bound ligand, the RMSF was calculated. The protein fluctuation features were essentially identical across all compounds, with the lowest fluctuating values detected in compounds 1–4, indicating that these compounds are more stable or have stronger binding and that more protein residues are fixed because of their binding with the ligands (Figure 4).
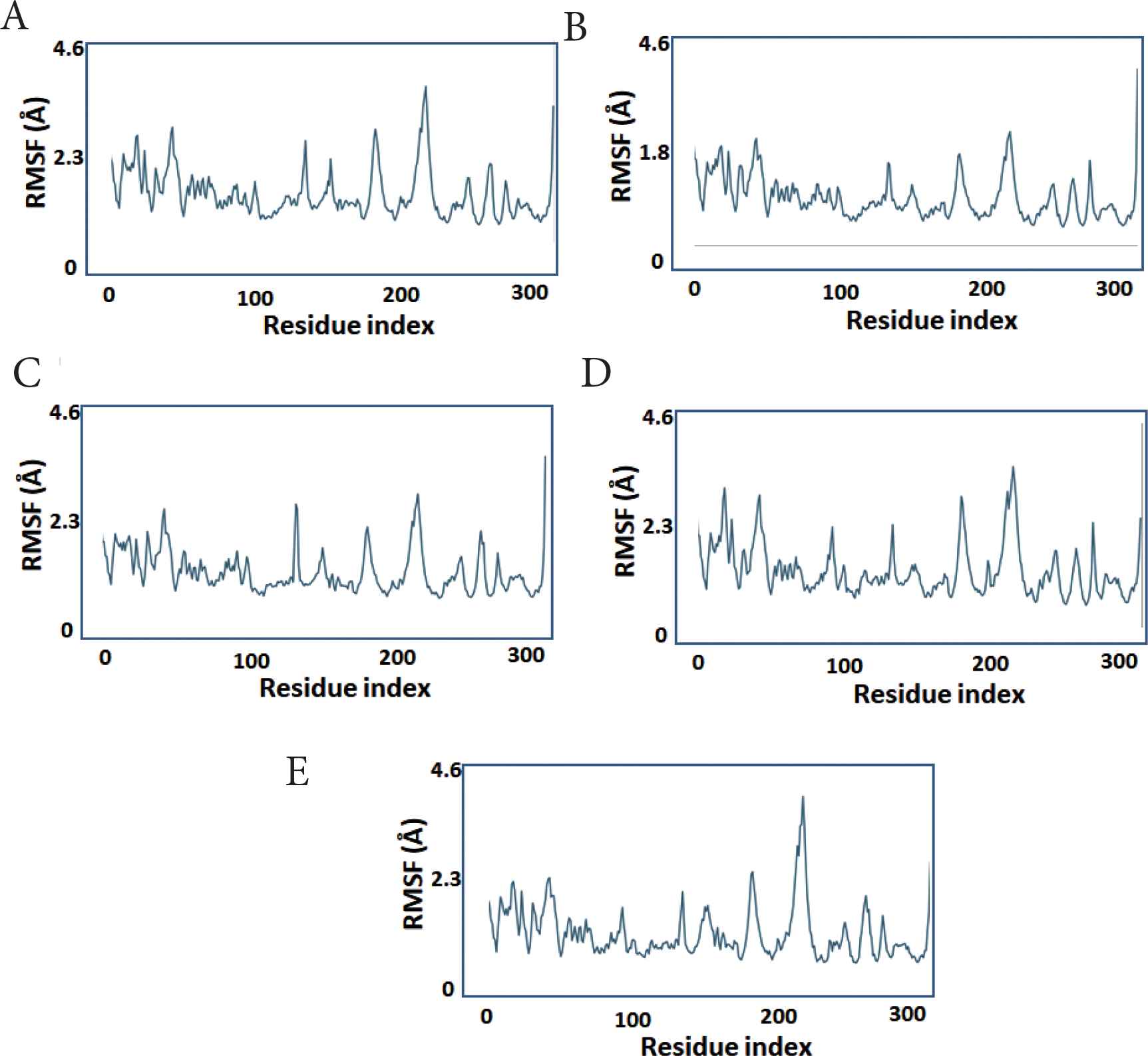
The protein per-residue RMSF during simulation. X-axis: PLpro residue number. Y-axis: estimated RMSF in Angstroms. (A) compound 1, (B) compound 2, (C) compound 3, (D) compound 4, (E) compound 5.
The Ligand RMSF (L-RMSF) was calculated to analyze the relative position of ligand atoms during simulation (Figure 5). The atom number is displayed on the X-axis, whereas the RMSF value is displayed on the Y-axis. The L-RMSF depicts how each ligand atom interacts with the protein and its potential entropic effects. Low L-RMSF was shown by compounds 2–4.
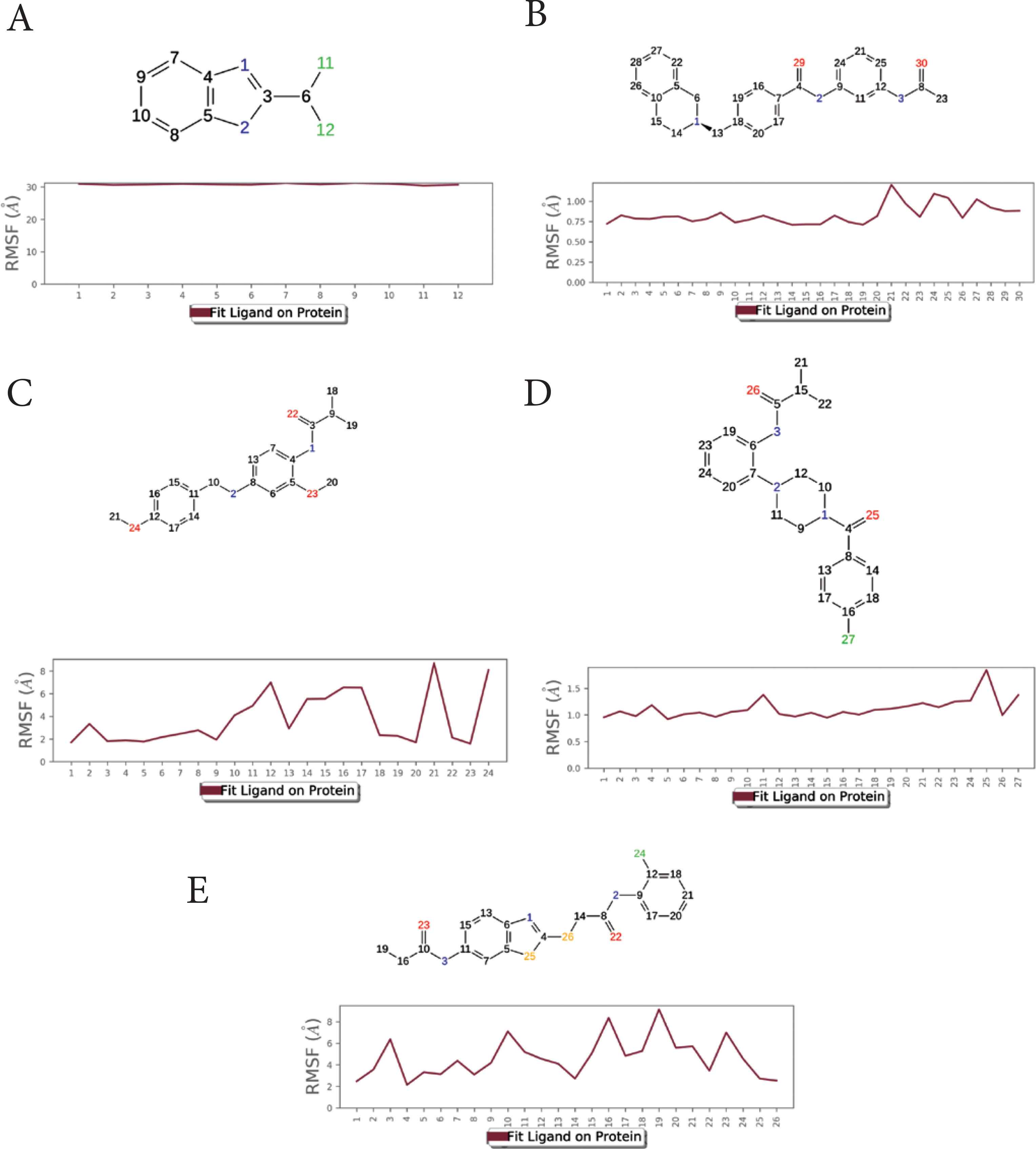
The ligand substructure RMSF during simulation. X-axis: atom number. Y-axis: estimated RMSF in Angstroms. (A) compound 1, (B) compound 2, (C) compound 3, (D) compound 4, (E) compound 5.
Protein–ligand contacts were monitored during the simulation. The traced contacts included hydrogen bonds, hydrophobic interactions, and ionic and water bridges (Figure 6). Most of the compounds formed ionic bridges, hydrogen bonds, or hydrophobic interactions with MERS-CoV PLpro. The major interactions were observed with the active site residues ASP1645 (H-bond), ASP1646 (water bridge), GLY1785 (H-bond), and PHE1750 (hydrophobic). Except for compound 5, all compounds produced at least two hydrogen bonds (Figure 7).

Dissection of interactions between each compound with the active site residues of MERS-CoV PLpro. (A) compound 1, (B) compound 2, (C) compound 3, (D) compound 4, (E) compound 5.
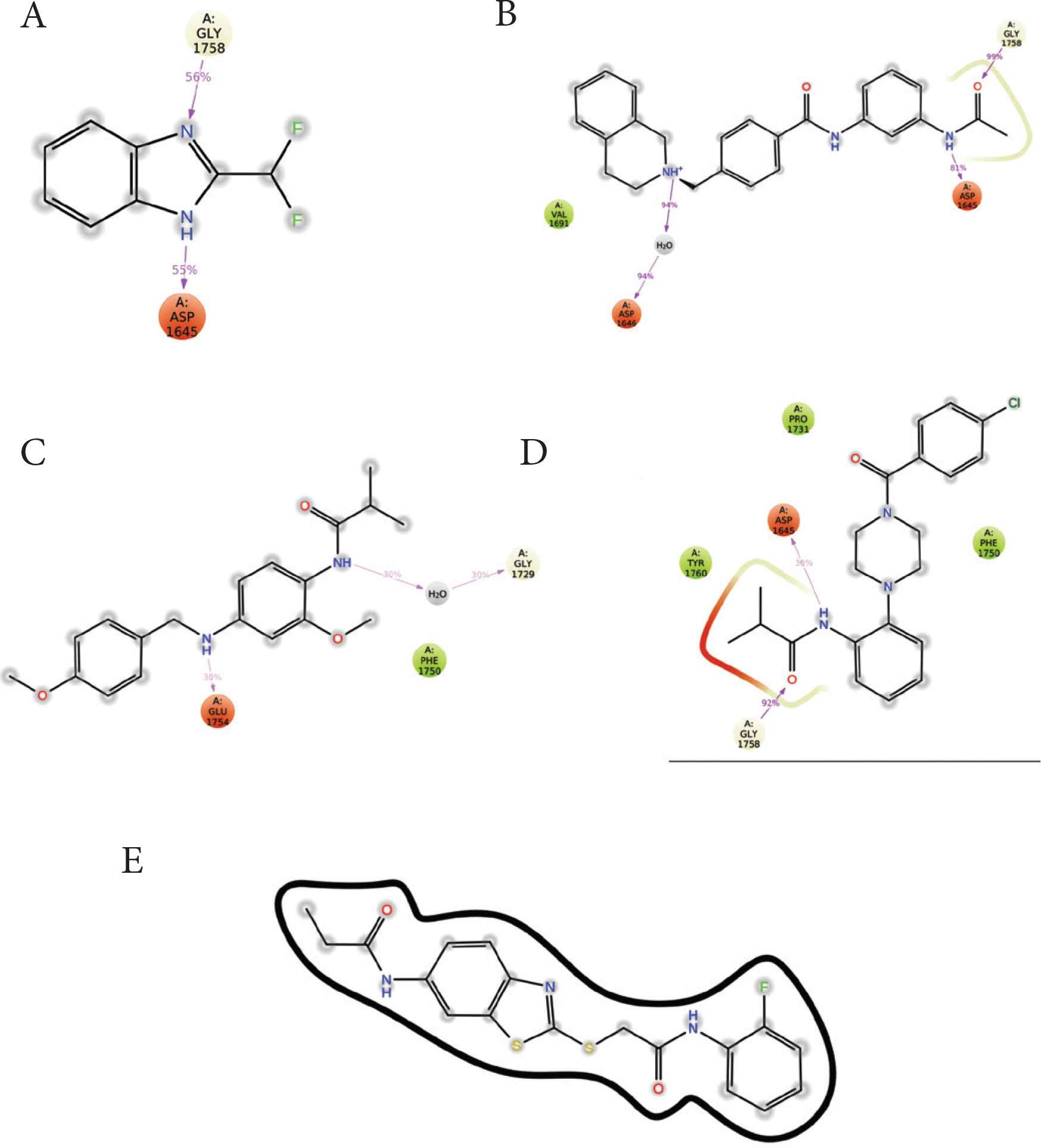
Ligand interaction maps of each compound with the active site residues of MERS-CoV PLpro. (A) compound 1, (B) compound 2, (C) compound 3, (D) compound 4, (E) compound 5.
The results of MM-GBSA are provided in Table 5. Compounds 2 and 4 produced the strongest binding by showing the lowest estimated MMGBSA_ΔG_Binding energy of −85.54 and −81.83, respectively.
| Time (ns) | C1 | C2 | C3 | C4 | C5 |
|---|---|---|---|---|---|
| 1 | −40.71 | −93.49 | −68.63 | −55.15 | −79.08 |
| 10 | −40.69 | −83.69 | −45.60 | −90.77 | −66.43 |
| 20 | −40.85 | −84.08 | −49.61 | −81.85 | −43.10 |
| 30 | 0.00 | −84.11 | −45.99 | −81.78 | −30.17 |
| 40 | −18.34 | −83.97 | −41.34 | −90.67 | −56.87 |
| 50 | −16.31 | −83.92 | −36.82 | −90.75 | −66.00 |
| AVG | −26.15 | −85.54 | −48.00 | −81.83 | −56.94 |
| SD | 17.21 | 3.90 | 11.02 | 13.78 | 17.72 |
AVG, average; SD, standard deviation.
Estimated MMGBSA_ΔG_Binding energy of compounds 1–5
4. DISCUSSION
Antiviral drug discovery is a vital research trend to control the spread of diseases caused by pathogenic viruses. Antiviral medications have been approved for a variety of viruses, for example, zidovudine, stavudine, abacavir, ritonavir, atazanavir, and enfuvirtide for HIV; acyclovir for human herpesvirus, and sofosbuvir for hepatitis virus [33]. Despite the serious sickness and deadly consequences of MERS-CoV infection, no approved medication to treat the virus has been produced.
Two compounds were identified as potential lead structures for further drug optimization studies. Compound 1 is a small-molecule benzimidazole derivative and showed the highest docking, enzyme inhibition and MERS-CoV plaque reduction. Previous studies also showed the potential inhibition of MERS-CoV PLpro by imidazole and purine derivatives [6]. Compound 4 is a piperazine derivative, reported here for the first time as a potential MERS-CoV inhibitor.
Lipinski’s rule of five or Pfizer’s rule of five was used to evaluate the drug-likeness of the top five compounds. This rule has been widely used to evaluate new compounds for drug-likeness and pharmacokinetics [34]. The rule states that orally active future drugs may violate at most one of the following rules: (1) no more than five hydrogen bond donors, (2) no more than 10 hydrogen bond acceptors, (3) a molecular mass less than 500 Da, and (4) an octanol–water partition coefficient (clog P) that does not exceed 5. All compounds showed no violations of the rule of five with favorable drug-likeness properties.
Compounds 1 and 2 occupied the active site of PLpro. The difluoroethane of compound 1 and butyraldehyde of compound 4 occupied the access to the active site residues (Figure 8). The ligand interaction maps revealed both hydrophilic and hydrophobic interactions between the compounds and MERS-CoV PLpro (Figure 9). Compound 1 formed stacking interactions with PHE1750 and two hydrogen bonds with ASP1645 and the side chain of GLY1758 (Figure 4A). Compound 4 formed a conserved hydrogen-bonding profile that was comparable to compound 1 except for the stacking interactions (Figure 9B). Despite both compounds’ close position and proximity to the MERS-CoV PLpro active site, no interaction was observed with the catalytic triad, composed of CYS1594, HIS1761, and ASP1776. MD simulation showed compounds 2 and 4 produced favorable protein conformation and stable protein binding and participated in various hydrogen bonding or hydrophobic interactions. All compounds produced favorable binding free energy, especially compound 4. Although compound 2 produced the highest binding free energy, it exhibited no antiviral activity. This observation might be attributed to other factors such as cell penetration or enzymatic hydrolysis.
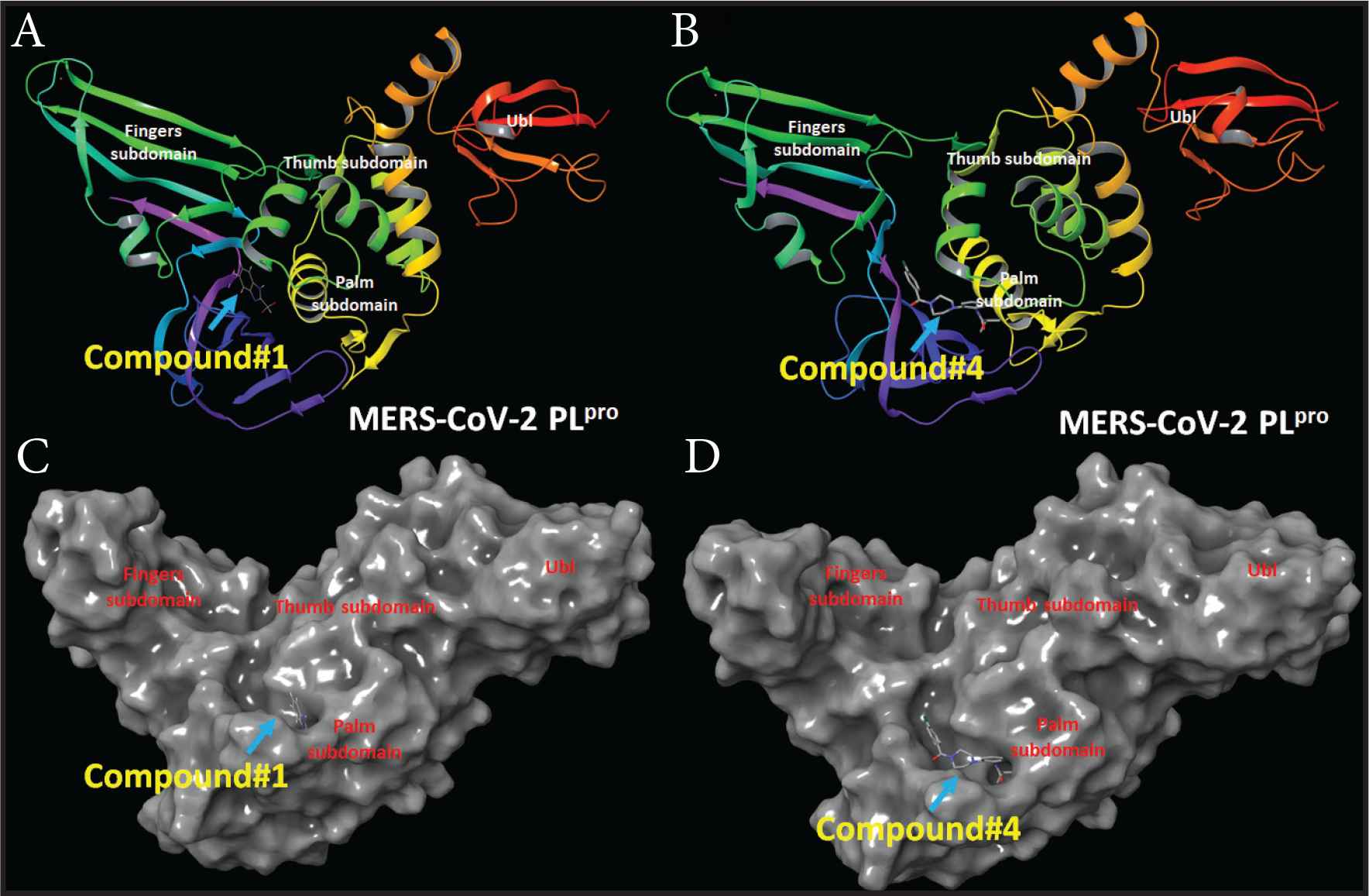
MERS-CoV PLpro structure and docking site of compounds 1 and 4. (A) MERS-CoV PLpro cartoon representation showing the site of compound 1. (B) MERS-CoV PLpro cartoon representation showing the site of compound 4. (C) Surface representation of MERS-CoV PLpro the docking site of compound 1. (D) Surface representation of MERS-CoV PLpro the docking site of compound 4.
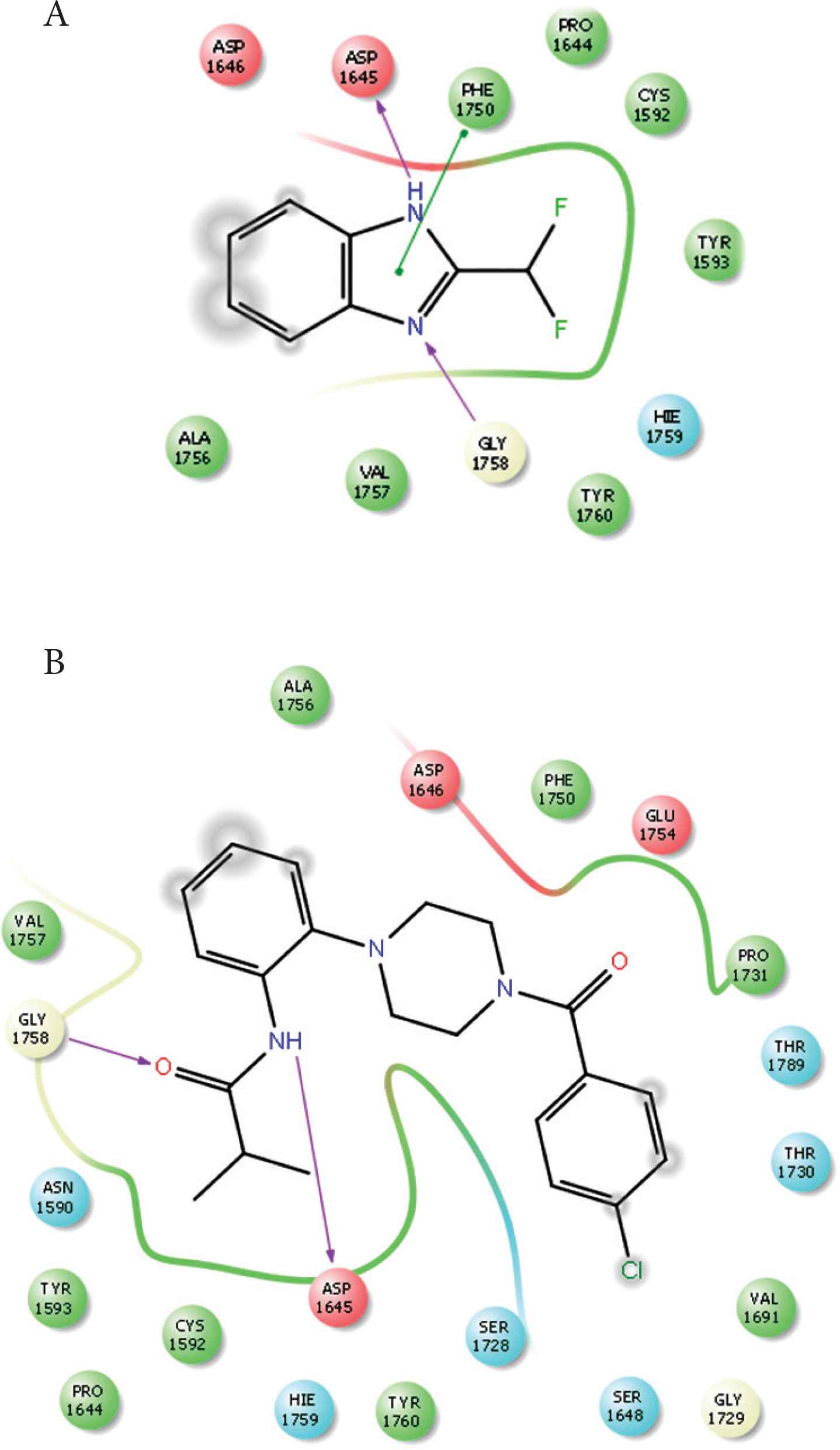
Ligand interaction maps. (A) Interaction profiles of compound 1 with MERS-CoV PLpro. (B) Interaction profiles of compound 2 with MERS-CoV PLpro. Hydrogen bonds are in purple arrows, stacking interaction in green sticks and negatively charged residues in red.
5. CONCLUSION
A structure-based approach was used to introduce new anti-MERS-CoV PLpro inhibitors, using virtual screening followed by biochemical and antiviral assays. The five compounds with the highest docking scores were found to inhibit MERS-CoV PLpro activity. The top two compounds reduced MERS-CoV plaque formation. Elucidating the mode of compound binding to the MERS-CoV PLpro active site revealed a lack of direct interaction of the compounds with the enzyme catalytic triad. Therefore, modifications of the two lead compounds obtained from this study to target binding with the catalytic triad might be a feasible approach for future drug discovery studies.
CONFLICTS OF INTEREST
M.K. received financial support from the excellence award 2020. The funding agent has no rule in the study design, results, or conclusions.
AUTHORS’ CONTRIBUTION
All authors: study conceptualization and writing (review and editing) the manuscript. MK and HJK contributed in data curation, formal analysis and writing (original draft). MK and HJK contributed in funding acquisition and project administration. MK, MAN, and HJK contributed in project supervision.
FUNDING
This work was funded by
ACKNOWLEDGMENTS
The authors acknowledge the Dr. Sulaiman Al Habib Excellence Award for the financial support, project number (COVID-19 RC15). Hyung-Joo Kwon was supported by grants from the National Research Foundation (2016M3A9B6916708) funded by the Ministry of Science and ICT in the Republic of Korea.
ETHICAL STATEMENT
This study did not use human or animals subjects.
Footnotes
REFERENCES
Cite this article
TY - JOUR AU - Mahmoud Kandeel AU - Byoung Kwon Park AU - Mohamed A. Morsy AU - Katharigatta N. Venugopala AU - Kentaro Oh-hashi AU - Mohammed Al-Nazawi AU - Hyung-Joo Kwon PY - 2021 DA - 2021/11/19 TI - Virtual Screening and Inhibition of Middle East Respiratory Syndrome Coronavirus Replication by Targeting Papain-like Protease JO - Dr. Sulaiman Al Habib Medical Journal SP - 179 EP - 187 VL - 3 IS - 4 SN - 2590-3349 UR - https://doi.org/10.2991/dsahmj.k.210921.001 DO - 10.2991/dsahmj.k.210921.001 ID - Kandeel2021 ER -