
Application of Deep Learning in Microbiome

- DOI
- 10.2991/jaims.d.201028.001How to use a DOI?
- Keywords
- Microbiome; Deep learning; Phylogeny
- Abstract
With the rapid development of high-throughput sequencing technology, massive microbial data has been accumulated. The understanding of the microbial data could help us to find the relationships between microbes and diseases. However, due to the high dimensionality, sparseness, and complexity of the data, traditional machine learning methods have insufficient learning and representational ability. Meanwhile, the rise of deep learning enables us to deal with these complex problems effectively. In this survey, we introduce the application of machine learning in microbial data analysis and focus on microbial classification and feature selection tasks. In particular, we discuss the current application and challenges of deep learning in microbial studies. Based on these discussions, we recommend that before using deep learning to conduct microbiome-wide association studies, it is essential to consider prior knowledge such as phylogeny, which would improve the accuracy and interpretability of the model.
- Copyright
- © 2020 The Authors. Published by Atlantis Press B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
A microbiota may consist of bacteria, archaea, fungi, viruses, and protists. All of the genetic materials in a specific ecological community are called the microbiome [1]. Recent studies in the microbiome have developed rapidly and researchers have found the microbiota could have a profound impact on human health. For example, studies have shown microbes in the human body have a great impact on immunology, digestion, absorption, and other physiological activities [2]. It is believed that there might be two-way communication between the brain and the intestine [3]. In addition, the intestinal flora may have a close relationship with irritable bowel syndrome [4] and it may lead to chronic kidney disease [5]. Therefore, the microbiome is often called the second human genome [6]. Compared with our human genome, it is much easier to intervene with the microbiota, which will make it an ideal target for medical treatment. However, it is estimated the number of microbial genes in the human body is much larger than the human genome. For example, the human gut contains more than 1 billion bacterium which have encoded more than 3 million genes [7]. Most of the species in the human body are concentrated in the intestinal tract, oral cavity, genital tract, and skin surface. However, the microbial communities in different environments are varying [8]. It is of great significance to understand the composition and function of the microbial community for different diseases or physiological states, which will greatly benefit the disease diagnosis and treatment [9].
However, the microbiome is complex, high diversity, and dependent. It is estimated there are more than 1000 kinds of bacteria in the human gut [10]. With the help of next-generation sequencing (NGS) technology, we could retrieve all genetic information in the sample [11]. NGS technology provides us with opportunities to understand the composition, function, and dynamic evolution of microbial communities, but it is often limited by metagenomic data analysis methods because the metagenomic data is too large and complex to be studied through visual means such as correlation analysis [12]. As a result, these methods need to guide new biological hypothesis or discovery from the massive data. To study the microbiome, researchers start to utilize machine learning methods to mine the relationships between microbes and their hosts. It is believed machine learning could learn and discover patterns from the data. However, the performance for each algorithm is usually dependent on feature engineering which is a process of using domain knowledge to extract features from the raw data. When performed manually, the process of feature engineering for machine learning can be prone to error. Besides, manual feature engineering is problem-specific—the algorithm cannot be applied to solve other issues. Another shortcoming of manual feature engineering is that the understanding of the problem limits it before thinking up so many features [13]. With automated feature engineering, none of these obstacles exist. Deep learning is a sub-discipline of machine learning and it automatically learns an end-to-end model from the data, eliminating the need for manual feature design. Therefore, deep learning could discover highly complex relevant features to improve prediction [14].
Deep neural networks (DNNs) are applied to conduct the microbial data analysis, but there are few surveys of deep learning in the microbiome rather than bioinformatics. In this survey, we will review related work in data analysis in microbiome studies, especially deep learning. Our survey will include four parts: first, we will give a brief introduction to the microbiome research background and its relationship with human health. Also, we will explain why it is time to apply machine learning and deep learning for microbial data analysis; second, we will review the research of machine learning in microbial data analysis; after that, we will discuss deep learning in microbial studies; lastly, we will explain some challenges of deep learning. The survey gives a snapshot of the field at present and it is naturally somewhat biased towards the authors' view, even though we hope that it provides useful information to the reader.
2. MACHINE LEARNING IN MICROBIOME
It is possible to understand better the hierarchical structure and composition of the microbial community via classifying microbial samples. The classification of microbial samples refers to identify microbial samples from different phenotypes in the environment [15]. One of the main goals of the microbial study is to explore the relative abundance of microbes, find the association between microbes and diseases, and analyze the different states of the disease to lay the foundation for further application in the follow-up treatment [9] or forensic identification [16].
The machine learning methods are used to conduct microbiome data analysis, including two steps: data preprocessing (such as feature selection) and model prediction (such as supervised learning). The general workflow of machine learning methods on microbiome data analysis includes (Figure 1): first of all, sequencing data needs to be collected from bacterial communities associated with various environments or hosts. What's more, these sequences can be directly used as input to a machine learning model, or they can be preprocessed. The preprocessing step usually improves the prediction results of the model. For example, it will be easier to construct a better prediction model if the model can learn the hierarchical relationship of the microbiome in advance. Finally, it may also improve the prediction of machine learning algorithms for the microbial associated disease after embedding structural information, such as the phylogenetic relationship among pedigrees and the average nucleotide similarity network between sequences.
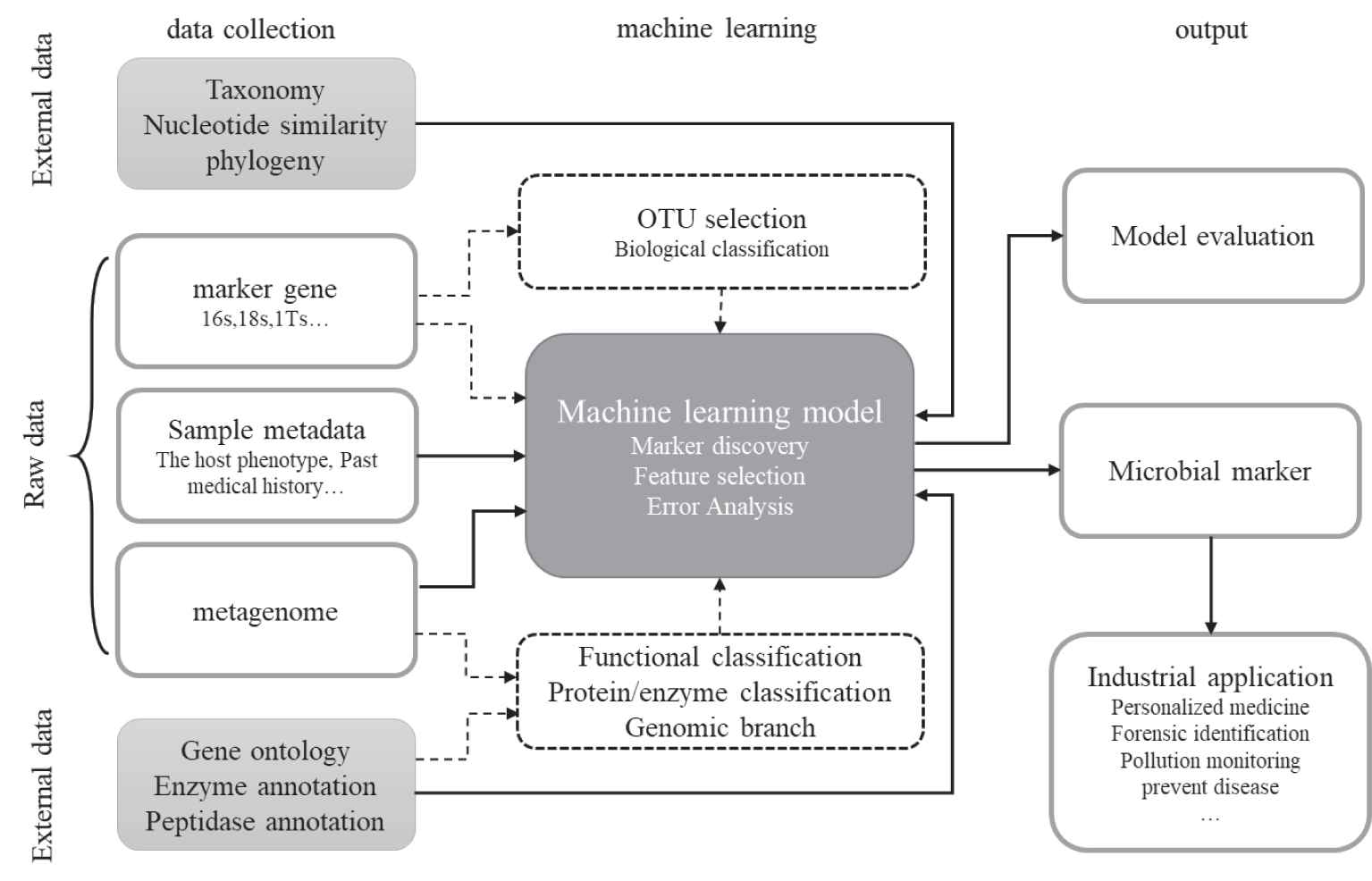
The general process of machine learning methods on microbial data analysis
Operational taxonomic units (OTUs) are the most common data representation for marker genes (16S rRNA genes) sequencing [17]. Knights et al. [15] benchmarked a classification task algorithm and found that the random forest (RF) achieved the best performance. Although the classification error of the elastic net (ENet) classifier was higher than that of RF, it was still helpful for feature selection as the preprocessing step of other classifiers. They suggested using a machine learning method to analyze the human body related to microbial composition and summarized the overall analysis process. Host-related microbial community composition was specific and closely related to diseases. Statnikov et al. [18] systematically compared 18 major machine learning methods in the classification task. They found the most efficient machine learning methods for accurately classifying sample data were SVM, Kernel Ridge Regression, and Laplace prior Bayesian Logistic Regression.
Machine learning could also be applied to detect microbial community composition and functional correlation analysis. Yazdani et al. [19] focused on the functional characteristics of the microbiome of inflammatory bowel disease to determine how microorganisms played a role in health and diseases. They developed and trained a two-step classifier to identify major changes in intestinal microbiome abundance between healthy and inflammatory bowel disease populations via the Kolmogorov–Smirnov (KS) test and RS. To determine the relationship between gut microbiota composition and clinical features of irritable bowel syndrome, Tap et al. [20] analyzed samples using L1 regularized logistic regression and found 90 OTUs that could be used to measure the severity of irritable bowel syndrome. Pasolli et al. [21] developed a machine learning analysis framework to assess the association between microbiome and phenotypes. They recommended using a RF approach based on species' abundance to predict diseases.
3. DEEP LEARNING IN MICROBIOME
Deep learning has achieved amazing success in areas such as image recognition, text processing, and automatic translation. Therefore, more and more researchers are trying to apply deep learning techniques to biomedical data analysis [22,23,24,25]. Compared with traditional machine learning, the advantage of deep learning lies in its end-to-end learning ability, which can automatically discover multiple representations to achieve better prediction. Among these deep learning architectures, low-level features (e.g., patterns in DNA sequence motifs or pathological images), high-level features (e.g., damaged mRNA splicing or asymmetric skin lesions), and output (e.g., cancer detection), all of these features can be learned from the data to reduce or eliminate the need for manual feature engineering [26].
Currently, with the development and application of high-throughput technology, a large number of microbiome data are emerging [27,28]. As a result, the deep learning method for metagenomics has attracted more and more attention. This section will focus on the following three applications of deep learning in microbial data analysis: metagenomic classification, metagenomic gene prediction, and microbiome-wide association studies (MWAS).
3.1. Metagenomic Classification
To characterize the diversity of the microbial community is one of the main objectives for metagenomic research. The classification and analysis of microbial sequencing reads are known as taxonomy, in which sequence reads are classified or clustered to specific bins [29,30,31]. Abe et al. [32] proposed an unsupervised neural network algorithm names self-organizing maps (SOMs) for de novo genome binning. SOM was used to analyze the dinucleotide, trinucleotide, and tetranucleotide frequencies in various prokaryotic and eukaryotic genomes. Essinger et al. [33] applied the adaptive resonance theory (ART) to cluster similar genome fragments and showed ART achieved better performance than k-means. Interestingly, methods based on interpolating the Markov model were better than these early genome binning techniques [34]. Liang et al. [35] reported a bidirectional long short-term memory (LSTM) with the self-attention mechanism named DeepMicrobes to conduct taxonomic classification for metagenomics, which could precisely identify species from microbial community sequencing data. In addition, Rojas-Carulla et al. [36] introduced a convolutional neural network (CNN) model for the shotgun metagenomic classification named GeNet. GeNet was trained from raw DNA sequences and exploited a hierarchical taxonomy between organisms via a novel architecture. However, neural networks were less frequently used for reference-based taxonomic classification because the training was time-consuming. TAC-ELM [37] was the first neural network-based method to classify massive amounts of metagenomic data, which introduced a new sequence composition-based taxonomic classifier via extreme learning machines. Fiannaca et al. [38] proposed a 16S short-read sequences based on k-means representation and deep learning architecture, in which a classification model was generated for each taxon (from phyla to genera).
It is believed that viruses play an important role in the microbial community [39]. However, the viruses are difficult to classify because they have not marker genes. It is essential to detect viral signals from the mixed metagenomic sequences. Fang et al. [40] presented a 3-class classifier named PPR-Meta, a CNN architecture, to identify both phage and plasmid fragments from metagenomic assemblies. Also, Ren et al. [41] developed a CNN model named DeepVirFinder to identify viral sequences in metagenomics, while VirNet used ‘deep attention’ [42], a technique commonly used for natural language processing. In addition, Tampuu et al. [43] proposed a CNN-based method named ViraMiner to detect viral contigs in diverse human biospecimens. ViraMiner was composed of the global max-pooling (pattern branch) and global average-pooling (frequency branch) after the convolutional layer. Kristopher et al. [44] introduced a hybrid approach to mining viral genomes named VIBRANT which uses neural networks of protein signatures from nonreference-based similarity searches with hidden Markov models (HMMs) as well as a v-score metric to maximize identification of diverse and novel viruses.
3.2. Metagenomic Gene Prediction
The NGS techniques used in metagenomics have generated many thousands of short reads. Gene recognition is a necessary step to fully understand the functions, activities, and effects of genes in cellular processes. Accurate identification of genes in metagenomic fragments is one of the fundamental problems for metagenomics [45]. In many situations, metagenomic reads are from thousands of highly heterogeneous species. Besides, high sequence coverage for a single species is often unavailable. It is difficult to assemble short reads into long overlapped contigs. One way is to bypass the assembly and find the genes directly from these short reads. Zhang et al. [46] proposed a deep stack network learning model named Meta-MFDL to predict metagenomic genes by fusing multiple characteristics of short reads, such as monoamine acid usage, ORF length coverage, and Z-curve features.
Meanwhile, bacterial antimicrobial resistance is usually genetically encoded and it is urgent to identify resistance genes in metagenomic samples [47]. Arango-argoty et al. [48] proposed DeepARG-SS and DeepARG-LS to determine the potential resistance gene, gene exchange hotspot, and diffusion pathway of a new antibiotic. Both DeepARG-SS and DeepARG-LS were constructed for short-read sequences and full gene length sequences, respectively. Due to the emergence of antibiotic-resistant bacteria, there is an alarming requirement to discover new antibiotics. Recently, Jonathan et al. [49] trained a DNN to predict antibiotics through the discovery of structurally distinct antibacterial molecules.
3.3. Microbiome-Wide Association Studies
MWAS are to predict the relationship between the microbiome and the disease state. MWAS are similar to genome-wide association studies. They aim to identify microbial species, genes, or metabolites associated with the disease phenotype [9,50,51], which often involve feature selection and classification.
In the metagenomic sample classification task, Ditzler et al. [52] tried two deep learning methods: deep belief network and recursive neural network, to determine whether the methods of DNNs were suitable for metagenomic analysis. They found traditional machine learning methods were powerful classifiers when the data was limited. Also, Nguyen et al. [53] proposed a method named MET2IMG to predict the disease state. To make it easy for CNNs, MET2IMG mainly utilized “filling” and t-SNE embedding methods to construct “synthetic images” from the metagenomic data. Since related organisms tend to have similar characteristics, the phylogeny is helpful to classify and infer ecological function, as well as a tool for organizing and understanding the microbial world [54]. Therefore, phylogenetic knowledge could improve the model's performance. Reiman et al. [55] introduced PopPhy-CNN to predict host phenotypes via metagenomic samples by embedding phylogenetic knowledge. Fioravanti et al. [56] introduced a new deep learning architecture, Ph-CNN, based on the CNN to conduct classification metagenomic samples. The ancestor distance defined on the phylogenetic tree and the sparse version of multi-dimensional scaling of Ph-CNN were used to embed the phylogenetic tree into the Euclidean space. Zhu et al. [57] proposed a deep forest that kept the spatial structure between nodes by embedding the phylogenetic tree to conduct the classification. Nathan et al. [58] conducted a comparison of methods, including tree-based (such as gcForest) and CNN-based (such as PopPhy-CNN) on microbial abundance features in different datasets.
Feature selection for MWAS is to discover the meaningful microbial biomarkers to guide the noninvasive diagnosis [59]. Many feature selection methods have been proposed for the microarray gene expression and mass spectrometry-based proteomics data to identify disease-associated genes or proteins [60]. When algorithms are applied to these high-dimensional data, a critical problem is known as the curse of dimensionality. Dimensionality reduction is one of the powerful ways to address the issue [61]. Autoencoder is a deep learning approach to learn latent representation to achieve this purpose [62]. However, autoencoder is considered as a feature extraction process. To deal with feature selection via deep learning, Zhu et al. introduced an ensemble feature selection method based on Deep Forest to conduct MWAS [57]. Compared to DNNs, Deep Forest is composed of decision trees and each decision tree could guide feature selection [63]. Meanwhile, Zhu et al. proposed a graph embedding approach to identify meaningful microbial biomarkers through deep feedforward neural network [64].
4. PROBLEMS OF DEEP LEARNING FOR MICROBIOME
The biological data is complex and hierarchical, it is not easy to get valuable information with simple analysis tools. Compared with traditional machine learning methods, deep learning has advantages on pattern discovering automatically. However, there are still many challenges for deep learning to conduct microbial data analysis [65].
4.1. Black Box
In the last decade, the application of DNNs to long-standing problems has brought a breakthrough in performance and prediction power [66]. However, high accuracy, deriving from the increased model complexity, often comes at the price of loss of interpretability, i.e., many deep models behave as black-boxes and fail to provide explanations on their predictions. While in specific application fields, this issue may play a secondary role in high-risk domains, e.g., health care or self-driving cars, it is crucial to building trust in a model and being able to understand its behavior. We are unlikely to trust a prediction if we do not understand how it was made. It is believed there are two major reasons why the interpretable model is vital for bioinformatics, not only microbial data analysis. First, understanding how predictions are made is essential to identify mistakes or biases in the input data when the model is trained. Second, deep learning could learn novel patterns from the massive biological data, which will guide meaningful insights if the model can be interpreted [67].
However, What makes it even worse is that there is no unified and standard definition for the interpretability of deep learning [68]. Techniques of interpretability have been adopted to a wide range of problems, and various meanings, such as understanding, interpreting, or explaining. For example, Grégoire et al. [69] gave an excellent review of methods to interpret and understand DNNs. In their survey, the authors focused on the post-hoc interpretability to understand what the model predicts (e.g., categories) in terms of what is readily interpretable (e.g., the input features) based on a trained model.
4.2. A Large Amount of Training Data
A large number of data is required to train DNNs. However, due to privacy concerns and high-cost issues (e.g., shotgun sequencing), it is impractical to obtain a large amount of biological data. Machine learning methods are superior to DNNs, such as XGBoost [70] when the data is limited. One of the main challenges to train DNNs without enough data is the risk of overfitting: when the training error is low while the test error is large, the models' generalization ability will become poorly. Fortunately, there are some approaches to alleviate the overfitting problem, such as Dropout [71]. Dropout will randomly remove neurons and their connections, which will reduce the capacity or thinning of the network during training. However, the overfitting problem is still one of the threats for the small biological or microbial data sets.
Furthermore, DNN requires a lot of computing resources during training, which is often computationally intensive and time-consuming and usually requires graphics processing units (GPUs) to process.
4.3. Model Selection and Hyper-Parameters Tuning
At present, there are many types of DNNs and the practitioners and researchers propose a growing number of new models. However, each model is varying in different scenarios, and it isn't easy and direct to choose a deep learning architecture for the specific task. Also, many hyper-parameters such as regularization degree, learning rate, number of neurons, etc., are required to tune and debug for the model to achieve optimal results. Therefore, efficiently choosing a suitable network architecture and fine-tune its hyper-parameters for a specific dataset is a time-consuming task given the staggering number of possible alternatives.
Automated machine learning (AutoML) is proposed, which is devoted to developing algorithms and solutions to enable people with limited machine learning background knowledge to use machine learning models easily [72]. In addition, many software libraries make the implementation of deep learning models easier. TensorFlow [73], Keras [74], CNTK [75], and Pytorch [76] are some examples of such libraries. Despite the availability of such libraries and tools, the tasks of picking the right neural network model and its hyper-parameters are usually complex and iterative. As a result, Steven et al. [77] proposed a method named multi-node evolutionary neural networks for deep learning (MENNDL), which was used to automate network selection on computational clusters through hyper-parameter optimization performed via genetic algorithms. Besides, automatic model selection (AMS) is a flexible and scalable method to automate the process of selecting artificial neural network models [78].
5. CONCLUSIONS
There are two basic questions to answer for the microbial studies, who's there and what they are doing. Many studies use the 16S rRNA gene as a taxonomic marker, then develop predictive models that can classify samples of disease states or habitats correctly. Compared with 16S rRNA gene sequencing, the shotgun metagenomics offers increased resolution, enabling a more specific taxonomic and functional classification of sequences as well as the discovery of new bacterial genes and genomes. Therefore, deep learning is preferred for the metagenomic data analysis, such as the classification or contig binning. But most of the current researches are focusing on different deep learning architectures(e.g., CNN or LSTM) and there is less work on deep transfer or reinforcement learning for the microbial studies.
This paper discussed deep learning for microbial data analysis, including metagenomic classification, metagenomic gene prediction. In particular, this survey introduced the application of deep learning in MWAS and further suggested a deep learning method based on the phylogenetic tree, which could improve the classification performance. We also analyzed the problems of deep learning in biomedical data analysis, including the model's interpretability, the need for a large amount of data and hyper-parameters tuning.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
AUTHORS' CONTRIBUTIONS
QZ, BH and XJ conceived the concept of the work. QZ and BL performed literature search. QZ, BH and HS wrote the paper. All authors have approved the final manuscript.
ACKNOWLEDGMENTS
This research is supported by the National Key Research and Development Program of China (2017YFC0909502) and the National Natural Science Foundation of China (No. 61532008 and 61872157).
REFERENCES
Cite this article
TY - JOUR AU - Qiang Zhu AU - Ban Huo AU - Han Sun AU - Bojing Li AU - Xingpeng Jiang PY - 2020 DA - 2020/11/04 TI - Application of Deep Learning in Microbiome JO - Journal of Artificial Intelligence for Medical Sciences SP - 23 EP - 29 VL - 1 IS - 1-2 SN - 2666-1470 UR - https://doi.org/10.2991/jaims.d.201028.001 DO - 10.2991/jaims.d.201028.001 ID - Zhu2020 ER -