
Distribution of hemoglobinopathy disorders in Saudi Arabia based on data from the premarital screening and genetic counseling program, 2011–2015
- DOI
- 10.1016/j.jegh.2017.12.001How to use a DOI?
- Keywords
- Hemoglobinopathy; Thalassemia; Sickle cell disease; Premarital screening; Saudi Arabia
- Abstract
The prevalence rates of β-thalassemia (β-thal) and Sickle Cell Disease (SCD) in Saudi Arabia are considered one of the highest compared to surrounding countries in the Middle East (0.05% and 4.50%, respectively). In this study, Secondary data analysis was obtained from the premarital screening and genetic counseling program (PMSGC), and included 12,30,582 individuals from February 2011 to December 2015. Prevalence rates (per 1000 population) for β-thal and SCD were calculated for carrier status, disease status and their combination. During the 5-year study period, the overall prevalence rate per 1000 population for β-thal was 13.6 (12.9 for the trait and 0.7 for the disease). The prevalence rate for SCD was 49.6 (45.8 for the trait and 3.8 for the disease). Rates for β-thal were found to decrease from 24.2 in 2011, to 12 in 2015. However, SCD rates remained rather constant and ranged from 42.3 in 2011 to 49.8 in 2015. The highest rate for both β-thal and SCD was observed in the Eastern and Southern regions. This result reflects major accomplishment of the PMSGC. This study recommends further improvement in preventive measures in high-risk regions, and enhanced community awareness to provide the highest rate reduction for these disorders.
- Copyright
- © 2017 Ministry of Health, Saudi Arabia. Published by Elsevier Ltd.
- Open Access
- This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
1. Introduction
Hemoglobinopathies, genetic disorders of hemoglobin, are the most common inherited disease in humans [1]. Global estimates indicate that approximately 3,00,000–4,00,000 infants are diagnosed yearly with hemoglobin disorders [2]. Two hemoglobinopathy disorders, thalassemia and sickle cell disease (SCD), have received significant attention of the global public health community due to their impact on increased mortality and morbidity among affected individuals [3,4]. Some forms of these disorders are considered serious autosomal recessive phenotype disorders, such as beta-thalassemia (β-thal) major, a lethal genetic disorder [5]. Yearly, approximately 240 million cases of heterozygous β-thal are diagnosed worldwide, mostly in the Mediterranean islands and in parts of Southeast Asia [6,7]. Worldwide SCD incidence in newborns was estimated to be 3,00,000 in 2008 [7]. SCD prevalence has been significantly increasing in most of sub-Saharan Africa, the Mediterranean, and the Middle East [7]. The prevalence rates of β –thalassemia (β-thal) and Sickle Cell Disease (SCD) in Saudi Arabia are considered one of the highest compared to surrounding countries in the Middle East (0.05% and 4.50%, respectively) [8].
Preventive screening programs have been adopted in several countries worldwide to reduce the prevalence of β-thal and SCD [9]. First, a premarital screening program was established in 1976 in Rome to determine the β-thal traits among students in a group of intermediate schools [10]. This was followed in 1980 by the establishment of an SCD preventive program in Virginia [1]. After this, many countries worldwide also adopted different types of preventive programs: premarital carrier screening, family-oriented approach to prevention, neonatal screening, antenatal screening for chromosome abnormalities and congenital malformations, and pre-implantation genetic diagnosis [11]. Countries that successfully implemented these programs included Italy, Greece, Cyprus, France, Iran, Thailand, Australia, Singapore, Taiwan, Hong Kong, and Cuba [9]. The goal of complete eradication for β-thalassemia was met in Cyprus, Italy, and Greece [12].
In the Middle East, eight countries have established premarital screening and genetic counseling programs (PMSGC), which constitute a mandatory step prior to receiving a marriage license and offer genetic counseling to couples at risk for hemoglobinopathy disorders [10]. Saudi Arabia introduced this program in 2001 and made it mandatory by 2004 [12]. A handful of studies have been conducted since 2004; they have shown a minor decline in the prevalence of SCD, but there were inconsistent reports about the prevalence of thalassemia. Six years after the launching the program, researchers have found a marked decrease in the number of at-risk marriages and have predicted a considerable reduction in the genetic disease burden in Saudi Arabia in upcoming years.
Overall, there is a great need for reducing the burden of hemoglobinopathy disorders, specifically the β-thal and SCD in Saudi Arabia. Having a successful mandatory premarital testing program will contribute to this target. Ongoing monitoring of trends in hemoglobinopathy disorders over time is necessary to evaluate the success of the PMSGC program in achieving the targeted reduction, and eventually elimination, of disease burden. In the light of this situation, this study was conducted to assess recent time trends in β-thal and SCD prevalence rates and their distribution by demographic characteristics and geographic regions in Saudi Arabia using data from the PMSGC program for the period of February 2011 to December 2015. Findings of this study will help health officials monitor trends in hemoglobinopathy disorders over time. This data will eventually contribute to ascertaining the effectiveness of current prevention and control efforts and provide information useful for policymakers interested in reducing the prevalence of hemoglobinopathy disorders.
2. Methods
2.1. Study design
A secondary data analysis was performed using data from the PMSGC program, housed within the Saudi Ministry of Health’s Genetic Department. Genetic screening data was obtained for all 12,30,582 individuals seeking to obtain a marriage certificate between the periods of February 2011 to December 2015. In February 2011, the program began computerized data entry.
Blood samples were taken, the results were shared with examinees, and genetic counseling was given. The database included demographic information as well as genetic test results for SCD and β-thal allowing classification of disease status positive, negative, or trait [1].
2.2. Data source – Premarital screening and genetic counseling program
Saudi Arabia launched its premarital screening and genetic counseling program in 2004. A royal decree issued in 2003 mandated premarital screening for the genetic diseases β-thal and SCD as a requirement to obtain marriage certifications [13]. Designated health centers were established for the program and equipped with medical supplies, personnel, and laboratory services [2]. There are currently 125 premarital health-screening centers across Saudi Arabia, all of which report data to the Ministry of Health (MoH), Department of Genetic Diseases [14]. The program offers free testing and counseling for couples looking to get their marriage certificate [3].
The PMSGC program not only identifies genetic blood diseases but also includes some infectious diseases such as hepatitis B, C and Human Immunodeficiency Virus. [13]. After these screenings, couples are provided with medical consultation in order to explain their chances of transmitting these diseases to their partner or future children, helping them plan healthy family outcomes [13].
For each partner in the health care center setting, the assigned program staff collects basic demographic information along with a medical history and general examination [13]. Hemoglobinopathy screening includes complete blood counts (CBC), peripheral blood film, reticulocyte count, high-performance liquid chromatography (HPLC) and sickling test for all the blood samples (in EDTA anticoagulant) [7]. The HPLC test is performed even if CBCs are normal. Several hemoglobinopathy disorders can be detected from this analysis such as β-thal, SCD, and different variations of hemoglobin-like HbC [4]. β-thal trait diagnosis is considered if a person has a Mean Corpuscular Volume (MCV) of <80 fL and/or a Mean Corpuscular Hemoglobin (MCH) of <27 pg, and a hemoglobin A2 level of >3.2% [15]. For diagnosis of the sickle cell trait, the test must show the presence of HbS with positive sickling [7]. Iron studies and serum ferritin, and DNA analysis are not routinely done along with the diagnosis of a-thal due to their low clinical significance in most of the cases [16,17].
2.3. Ethical considerations
The dataset was obtained from the data source in de-identified format. No patient identifiers were included, and a unique ID was used for each case in the dataset. A protocol was followed to protect the privacy and rights of patients. The study team fulfilled administrative and ethical approval requirements. This study was granted review exemption status by the Institutional Review Board (IRB) of Emory University.
2.4. Study variables
The variables included in this study were based on the 2011 study [8]. The dataset included the following variables: a unique identifier to differentiate each individual in the data set, city, age (years), gender (male/female), test results for β-thal major, β-thal trait, SCD, and SCT (Positive/Negative), year of testing and doctor notes.
2.5. Data management
Data was received as separate files with multiple lines per person reflecting each disease result for each individual, one file containing the positive test results and eight files containing the negative test results in the raw datasets, which there were approximately 1,54,336 observations in the positive file, and almost 75,22,356 observations in the negative files. Data was received in Excel format and analyzed using SAS 9.4 (SAS Institute Inc., Cary, NC). All subsequent data management, handling and statistical analyses were done using a terminal-end connection to a High Performance Computing Cluster at Emory University for further analyzing the data and saving results.
The SAS dataset was thoroughly examined for integrity, inconsistencies, inaccuracies, and invalid entries and changes were made to correct data errors and recode variables as needed. “Region” was recoded from its original 20-level variable representing health districts to a 13-level variable representing administrative districts, because population counts (denominators) were only available for administrative regions in Saudi Arabia. Saudi Arabia is divided into thirteen administrative regions: Al-Jouf, Asir, Baha, Eastern region, Hail, Jizan, Maddinah, Makkah, Najran, Northern Borders, Qasim, Riyadh, and Tabouk These regions are further subdivided into twenty health regions according to Ministry of Health [18]. Reclassifications in the variable “City” were made to be consistent with the changes made in “Region”. Tests for disorders/diseases not relevant for our study such as α-thal were removed [7]. Entries outside the research timeframe were removed. When a disease was labeled ‘other’, the test result and notes were examined for any relevant information that might be important for our study. For a group of subjects whose test result was entered as “see comment”, notes were examined to identify information that may inform “disease status” assignment. A total of 4588 individuals were described as possibly having either α-thal trait, iron deficiency, or silent β-thal or undetermined, RBC count, Hb, MCV, MCH, Hb A2, RDW, and Hb F in the β-thal traits, undetermined β-thal or can’t excluded in doctor comments [16]. From these results, confirmed cases of β-thal trait were 653.
All datasets were separately cleaned and transposed by unique identifiers to create a one-line per person data structure and subsequently merged using SAS PROC SQL. Variable names were altered to retain the original source of common variables to check for inconsistencies and ensure completeness.
Duplicate entries in the negative datasets were examined, and the latest entry per person was retained. Of 15,971 subjects from the negative data set, 770 had positive test results for β-thal, from which 768 matched the test result in the positive data set and 15,126 had β-thal trait, from which 15,124 matched the test result in the positive data set.
Of 59,326 subjects from the negative data set, 188 had positive test results for SCD, from which 90 matched the test result in the positive data set and 2243 had carrier test results for SCD, from which 1868 matched the test result in the positive data set.
Subjects with positive test results were deemed as positive even if they had a contrasting entry in the negative dataset.
All files were merged to create a final dataset that included 12,30,582 observations with the following set of variables for each observer: subject identifier, gender, age, city, disease results (β-thal major, β-thal trait, SCD, and SCT), doctor notes, and year of testing.
2.6. Statistical analysis
An exploratory analysis of the data was done and summary statistics for all independent variables were derived. Continuous variables were summarized with descriptive statistics (N, mean, standard deviation, range). Categorical variables were summarized with frequency counts and percentages within each category or between levels of a category as appropriate.
The prevalence rate and 95% confidence interval (CI) of β-thal and SCD overall, by year from 2011 to 2015, and for each region was estimated. Prevalence rates were calculated separately for disease and carrier statuses within each condition and estimated as: the number of affected individuals divided by the number of tested individuals per 1000 population [19]. Statistical significance was determined at the 0.05 level using two-sided P-values. Prevalence rates were compared using prevalence rate ratios and 95% confidence intervals (CIs) [19].
3. Results
3.1. Population characteristics
The overall study population of 12,30,582 individuals attended the premarital screening centers distributed across the 13 administrative regions in Saudi Arabia between February 2011 and December 2015. Approximately 49.7% were male with an average age of 27.8 years and (SD 8.85 years) and 50.34% were female with an average age of 22.6 years (SD = 6.4). Furthermore, a higher proportion of program attendees was in the 18–25 age group; represented as 46.2% for men and 53.8% for women (Table 1) [16]. The population distribution over the years is outlined in (Table 2) [20].
| Age groups (years)* | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gender | % | Age Range | Mean age ± SD | <18 (%) | 18–25 (%) | 26–35 (%) | 36–45 (%) | 46–55 (%) | >55 (%) |
| Males | 49.7 | 17–105 | 27.8 ± 8.9 | 7.6 | 46.2 | 64.4 | 66.1 | 83.8 | 24.1 |
| Females | 50.4 | 13–112 | 22.6 ± 6.4 | 92.4 | 53.8 | 35.7 | 34 | 16.2 | 5.3 |
[1] Belhoul KM, Abdulrahman M, Alraei RF. Hemoglobinopathy carrier prevalence in the United Arab Emirates: first analysis of the Dubai Health Authority premarital screening program results. Hemoglobin 2013;37(4):359–368. doi: 10.3109/03630269.2013.791627. PubMed PMID: 23647352.
Statistically significant association [1].
Age distribution for men and women attending premarital screening in Saudi Arabia, 2011–2015.
| Population screened | Β-thalassemia Trait | Sickle Cell Trait | |||||
|---|---|---|---|---|---|---|---|
| Year | 12,30,582 | Positive test | PR* | 95% Confidence Interval | Positive test | PR | 95% Confidence Interval |
| 2011 | 78,072 | 1892 | 24.2 | 23.2–25.3 | 3304 | 42.3 | 41.0–43.8 |
| 2012 | 2,58,581 | 4557 | 17.7 | 17.1–18.2 | 13,055 | 50.5 | 49.7–51.3 |
| 2013 | 2,68,097 | 2837 | 10.6 | 10.2–11.0 | 13,406 | 50.0 | 49.2–50.8 |
| 2014 | 2,76,236 | 2666 | 9.7 | 9.3–10.0 | 13,490 | 48.9 | 48.0–49.7 |
| 2015 | 3,05,871 | 3657 | 12.0 | 11.6–12.4 | 15,307 | 50.0 | 49.3–50.8 |
[2] Incebiyik A, Genc A, Hilali NG, Camuzcuoglu A, Camuzcuoglu H, Kilic A, Vural M. Prevalence of beta-thalassemia trait and abnormal hemoglobins in Sanliurfa Province in southeast Turkey. Hemoglobin 2014;38(6):402–404. doi: 10.3109/03630269.2014.978008. PubMed PMID: 25405917.
p-value <0.0001 for comparisons of PRs for β-thalassemia carrier by year [2].
Prevalence rates (PR per 1000) for β-thalassemia Trait and Sickle Cell Trait in Saudi Arabia form 2011 to 2015.
3.2. Prevalence rate, time trends and distribution of β-thal
Overall, for the 5-year study period, the estimated prevalence rate for β-thal obtained from this study was 13.6 per 1000; 12.9 per 1000 (N = 15,351 individuals) for the trait and 0.7 per 1000 (N = 787 individuals) for β-thal major. Among the total number of people examined, there were 11,74,181 individuals with a negative result for β-thal. The prevalence rate for the β-thal trait decreased from 24.2 per 1000 in 2011 to 12 per 1000 in 2015 (Table 1) [16]. The prevalence rate for β-thal major varied from 1 in 2011 to 1.6 in 2015. β-thal prevalence varied by regions where the highest rate was observed in Jazan region: 32.1 per 1000 [95%;CI30.8–33.4] for β-thal trait and 0.6 per 1000 [95%; CI 0.5–0.8] for β-thal major (Table 3, Fig. 3) [7]. The second highest rate was reported in the Eastern region with (23.7 per 1000 [95% CI; 23.1–24.4]) for β-thal trait and (0.4 per 1000 [95% CI; 0.3–0.5]) for β-thal major (Table 3, Fig. 3) [7]. Other regions showed rates that varied from 3 to 15 per 1000 for both β-thal trait and disease status. While β-thal rates showed a fluctuating pattern across regions by year, there was a general trend of decreased rates from 2011 to 2015 across the different regions, except for the Northern region (Fig. 1).
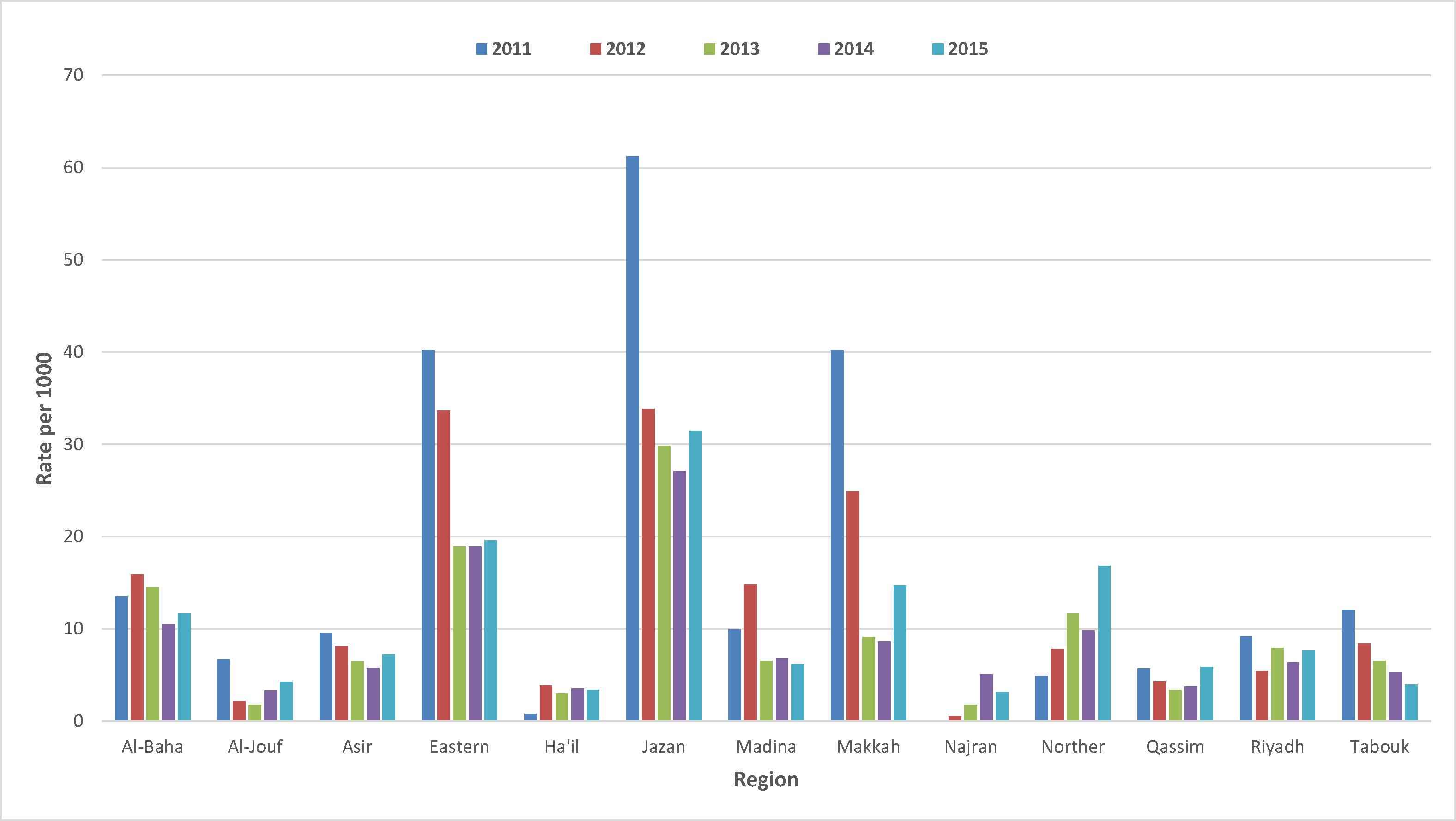
Prevalence Rates of β-thalassemia° by regions and time, Kingdom of Saudi Arabia, 2011–2015*. (°) β-thalassemia rates trait and disease status per 1000 are presented in a fluctuating pattern across 13 regions over a 5-year period. (*) P-value <0.0001 with the prevalence rate of β-thalassemia.
| β-thalassemia | Sickle Cell Disorder | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Regions | Population | Trait | 95%CI | Disease* | 95%CI | Carrier | 95%CI | Disease* | 95%CI |
| Al-Baha | 29,161 | 13.2 | 11.9–14.6 | 0.3 | 0.2–0.6 | 34.9 | 32.8–37.0 | 2.8 | 2.2–3.5 |
| Al-Jouf | 29,339 | 2.9 | 2.3–3.5 | 0.3 | 0.1–0.5 | 2.6 | 2.0–3.2 | 1 | 0.7–1.4 |
| Asir | 1,65,316 | 6.7 | 6.3–7.1 | 0.2 | 0.1–0.2 | 42.5 | 41.5–43.4 | 7 | 6.6–7.4 |
| Eastern Region | 2,11,727 | 23.7 | 23.1–24.4 | 0.4 | 0.3–0.5 | 114.4 | 113.0–115.8 | 9.8 | 9.4–10.2 |
| Hail | 38,567 | 3.3 | 2.8–3.9 | 0.00 | 0.0–0.1 | 2.0 | 1.6–2.5 | 0.1 | 0.0–0.2 |
| Jazan | 72,420 | 32.1 | 30.8–33.4 | 0.6 | 0.5–0.8 | 135.7 | 133.2–138.2 | 6.8 | 6.2–7.4 |
| Makkah | 2,38,978 | 14.4 | 13.9–14.9 | 1.9 | 1.7–2.1 | 31.3 | 30.6–32 | 1.9 | 1.7–2.1 |
| Maddinah | 81,286 | 8.1 | 7.55–8.79 | 0.2 | 0.1–0.3 | 13.7 | 13–14.6 | 0.8 | 0.6–0.9 |
| Najran | 24,836 | 2.4 | 1.9–3.1 | 0.00 | 0.0–0.2 | 12.6 | 11.3–14.1 | 0.3 | 0.2–0.6 |
| North Border | 22,260 | 7.6 | 6.5–8.8 | 3.4 | 2.7–4.3 | 4.0 | 3.3–5 | 0.4 | 0.2–0.8 |
| Qasim | 70,057 | 4.0 | 3.6–4.5 | 0.4 | 0.3–0.6 | 2.5 | 2.2–2.9 | 0.3 | 0.2–0.5 |
| Riyadh | 2,00,652 | 7 | 6.6–7.4 | 0.2 | 0.2–0.3 | 18.1 | 17.6–18.7 | 1.1 | 0.9–1.2 |
| Tabouk | 45,983 | 6.3 | 5.6–7.1 | 0.2 | 0.1–0.4 | 27.5 | 26.0–29.0 | 1.0 | 0.8–1.4 |
95%CI = 95% confident interval.
[3] Memish ZA, Owaidah TM, Saeedi MY. Marked regional variations in the prevalence of sickle cell disease and beta-thalassemia in Saudi Arabia: findings from the premarital screening and genetic counseling program. J Epidemiol Global Health 2011;1(1):61–68. doi: 10.1016/j.jegh.2011.06.002. PubMed PMID: 23856375.
Significant presentation among the B-thalassemia and Sickle cell disease p-value = 0.0001 [3].
Prevalence rate for β-thalassemia and Sickle Cell disorders by region, in Saudi Arabia, 2011–2015.
3.3. Prevalence, time trends and distribution of SCD
A higher prevalence rate was observed for SCD than β-thal; the overall rate was 5 per 1000 (N = 56,292) for the SCT and 0.38 (N = 4632) for SCD. A total of 11,69,408 screened negative for SCD. The total number of sickle tests done from 2011 to 2015 was 12,30,332, where the prevalence rate of SCD remained rather constant from 2011 to 2015 with an average rate 48.34 per 1000 (Table 1) [16]. Makkah was having the highest number of tests done, followed by the Eastern region and Riyadh. The highest rate of SCT was present in Jazan: 135.7 per 1000 [95% CI; 133.2–138.2]. The second highest rate was observed in the Eastern Region: 114.4 per 1000 [95% CI; 113.0–115.8] (Table 3, Fig. 3) [7]. In Asir, Al-Baha, Makkah rates ranged from 31 to 42 per 1000. The lowest rate for SCT was observed in Hail (2.0 per 1000 [95% CI; 1.6–2.5]) (Fig. 3). SCD was most prevalent in the Eastern Region (9.8 per 1000 [95% CI; 9.4–10.2]), followed by Asir and Jazan regions with their rates ranging between 6.8 and 7 per 1000. The lowest rate was present in Hail (0.1 per 1000 [95% CI; 0.0–0.2]) (Fig. 2). Within each region, rates of SCD were rather constant across the 5 years (Fig. 2).
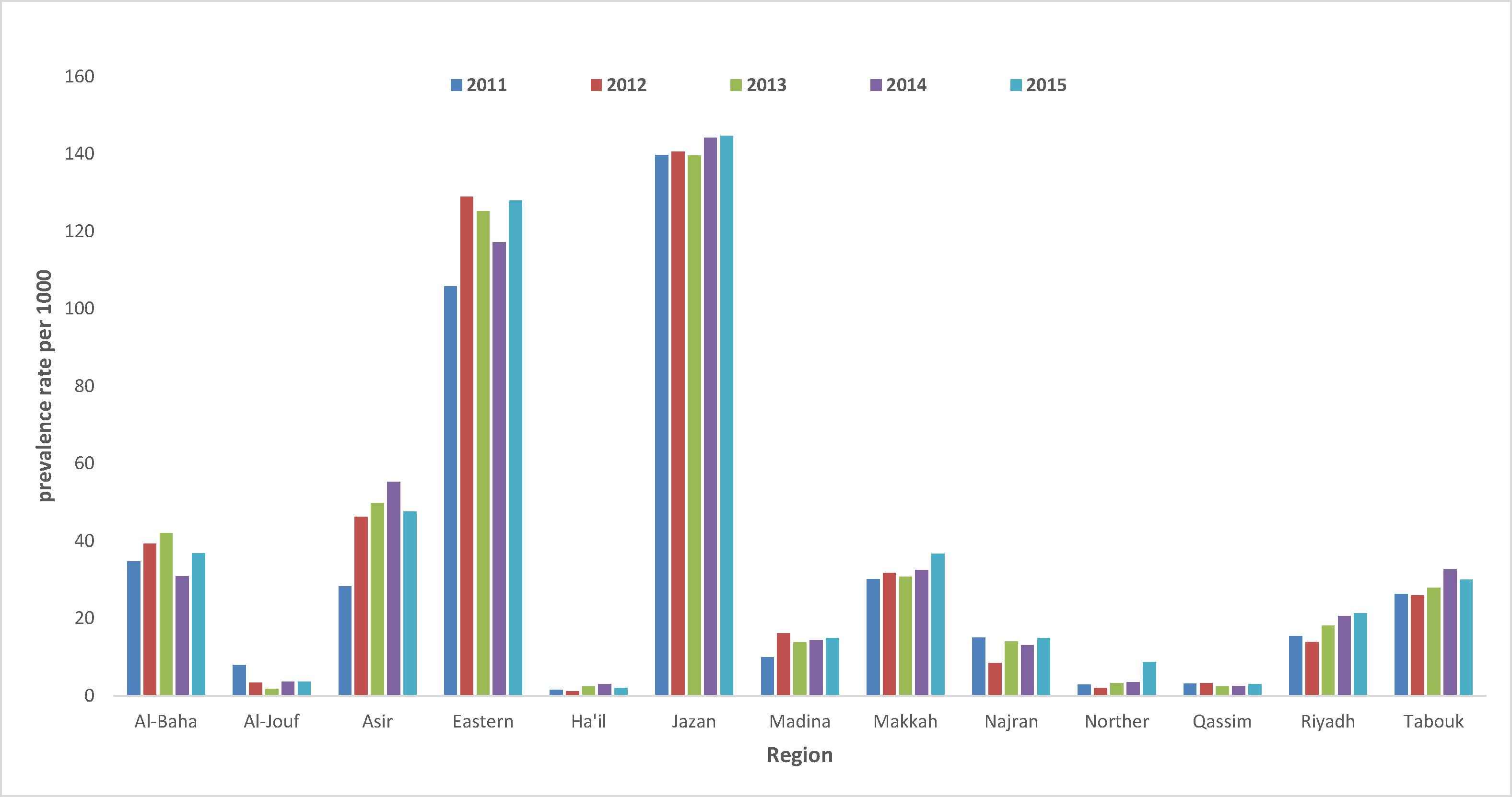
Prevalence Rates of high-risk presentation of Sickle Cell Disease by regions and time, Kingdom of Saudi Arabia, 2011–2015*. Sickle Cell Disease rates (trait and disease) per 1000 are presented in a fluctuating pattern across 13 regions over a 5-year period. (*) P-value <0.0001 with the prevalence rate of β-thalassemia.
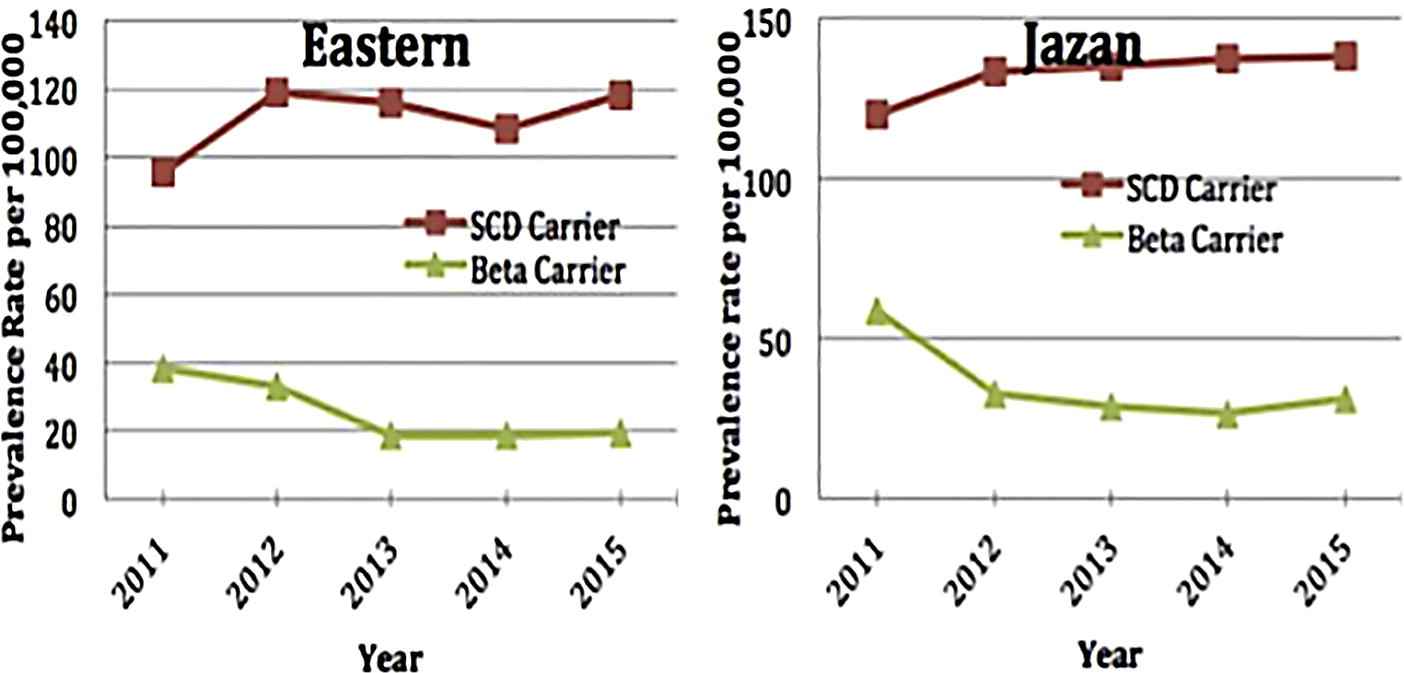
Prevalence rates for β-thalassemia and SCD carriers over time in the highest-rate regions**, Kingdom of Saudi Arabia, 2011–2015 (**) Eastern region and Jazan.
4. Discussion
This study assessed the prevalence, 5-year time trends, and distribution of β-thal and SCD in Saudi Arabia using data from the Saudi premarital screening and genetic counseling program from February 2011 to December 2015. A decreasing trend was observed in the prevalence of β-thal across the years; however, the prevalence of SCD remained rather constant.
Since the launch of the program, only one study published in 2011 has analyzed trends from 2004 to 2009. There were no available studies for comparison of the rates of β-thal and SCD post 2009.
Thalassemia is the most common genetic disorder in the world; the highest percentage of hemoglobinopathy disorders in KSA have been attributed to α-thal [21]. Up to 45% of the population in the Eastern region of KSA is heterozygous for α-thal [22]. However, because no biochemical diagnostic test is available for detection of α-thal carriers, and because red cell morphology may be quite reasonable in hemoglobin electrophoresis [22,23], the premarital screening program does not include this condition; therefore, it was not part of our study.
On the other hand, β-thal disorder accounts for the other portion of overall thalassemia prevalence. In our study, 1.4% of the study population had β-thal disease and was considered high-risk, as defined by the PMSGC program. The majority of these cases were β-thal carriers and the remaining had β-thal major. This result is similar to a 2004–2006 study that analyzed the first three years of data from the screening program [12]. Though the disease proportions were similar, the carrier numbers were distinct. Compared to the previous study which reported a rate of 3.2% for β-thal carrier status, this study observed a rate of 1.3% [12].
Studies from other Arab countries showed that the frequency of β-thal trait in general falls within the range of 2.0–10.0% [16]. Our study reported regional variations within Saudi Arabia in β-thal major. The regions of highest prevalence are arranged geographically along a belt from west to east, starting from the Western region (Baha, Jazan, and Makkah) toward the Eastern region [7].
In this study, SCD had the highest prevalence of the two hemoglobinopathy disorders. The overall prevalence was 5% and the majority of cases were SCT. The number of cases was rather constant over the course of the study period. The number of SCD and SCT cases per year was 1,38,145. The current prevalence increased slightly compared to that reflected in previously published studies among the same population; the SCD rate went from 3 per 1000 in a previous study to 4 per 1000 in this one, and the SCT rate went from 42 per 1000 in a previous study to 46 per 1000 in this one [7]. This increase may be explained by an increase in the disease survival rate attributable to improvements in health care services and utilization, so that more individuals with SCT or SCD are reaching marriage age [12,24].
Comparing the regional spread of SCD and β-thal, this study found that the highest prevalence for both occurred in the Eastern region and Jazan region, which is similar to the distribution reported in the 2004–2011 PMSGC study.
An important risk factor affecting the occurrence of both disorders, β-thal and SCD, is consanguineous marriage. Consanguinity is distributed in different rates around regions in Saudi Arabia [7]. Public health intervention approaches need to take these regional variations into consideration when implementing health promotion and healthcare services.
Malaria endemicity is reported to correlate with SCD occurrence. This study reflects this pattern; the regional prevalence of SCD is consistent with the regional distribution of malaria, which is also reported to be highest in Jazan. Having SCD is known to confer a survival advantage and a protective effect against malaria. Those with SCD in malaria-endemic areas have a prolonged life expectancy and better survival rate for this disorder [7,23].
One of the overall targets of the PMSGC program is to reduce and even eliminate the presence of β-thal and SCD in newborns. However, achieving this target is dependent upon the presence of other preventive programs such as prenatal screening and abortion, the implementation of which is restricted by ethical, religious and societal values [13,16].
One strength of the Saudi PMSGC program is the fact that it was made mandatory for all individuals seeking to get married. This will ensure increasing community awareness of hemoglobinopathy diseases, which will increase the efficacy of this program. This program has wide coverage involving 4% of the total population every year, and provides a valuable data resource and research opportunities. Besides determining the change in trends of these disorders, the data contributes to a more accurate assessment of the magnitude of hemoglobinopathy disorders in Saudi Arabia.
This study had some limitations. The study would have required a longer timeframe to observe the genetic consequences of the PMSGC program [25,26]. This study did not have access to data on the outcomes of the genetic counseling offered to those at risk of transmitting blood disorders to their offspring, so this study was not able to determine which couples decided to marry, and therefore this factor was not included in analysis of the PMSGC program [8]. Another limitation concerns the sample. Though our sample size was large, estimates for smaller regions may not be stable due to low counts.
A few recommendations emerged from our study to improve the effectiveness of the PMSGC program and lower the prevalence of hemoglobinopathy disorders. First, β-thal and SCD rates in newborns should be assessed as one component of evaluating the effectiveness of the premarital screening program. Second, health promotion and disease prevention efforts should be improved for those at higher risk of transmitting blood disorders. There should be a follow-up program post marriage to ensure a sufficient level of awareness. In addition, health education should be focused on younger age groups to improve their attitudes and receptivity towards genetic counseling. Third, higher risk regions should be targeted for the establishment of health campaigns that will increase the population’s level of awareness about the disorders and how serious they are. The media should convey messages around the risks of consanguinity. Fourth, more effective genetic counseling and psychological support should be provided before marriage to ensure that at-risk couples have a comprehensive awareness of risks before they reach a decision about getting married.
In conclusion, this study extends previous studies by providing an update on the recent trend in the occurrence of β-thal and SCD over a 5-year period. It revealed a marked reduction in β-thal but a steady state in sickle cell disease. This data will set the stage for continued monitoring of hemoglobinopathy disorders and will facilitate future evaluations of the effectiveness of existing preventive programs. This data yields important information for action for decision makers that will enable them to prioritize new programs and implement policy changes.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgment
Prima facie, I am grateful to God for giving me deep faith, good health and wellbeing that were necessary to complete this study. Besides my advisor, my deepest gratitude to Eng. Mohammed Suwaimil, for his support, continuous fellow up, patience and primary editor role. My sincere thanks to Dr. Abdullah Asiri and Dr. Scott McNabb for their guidance, Natalie Schulhofer and Yasmin Zaki for their outstanding review, Prabhjyot Saini for his great works in data analysis.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.jegh.2017.12.001.
References
Cite this article
TY - JOUR AU - Eman S. Alsaeed AU - Ghada N. Farhat AU - Abdullah M. Assiri AU - Ziad Memish AU - Elawad M. Ahmed AU - Mohammad Y. Saeedi AU - Mishal F. Al-Dossary AU - Hisham Bashawri PY - 2017 DA - 2017/12/15 TI - Distribution of hemoglobinopathy disorders in Saudi Arabia based on data from the premarital screening and genetic counseling program, 2011–2015 JO - Journal of Epidemiology and Global Health SP - S41 EP - S47 VL - 7 IS - Supplement 1 SN - 2210-6014 UR - https://doi.org/10.1016/j.jegh.2017.12.001 DO - 10.1016/j.jegh.2017.12.001 ID - Alsaeed2017 ER -