
Deep Learning Methodologies for Genomic Data Prediction: Review
 , Steve A. Adeshina
, Steve A. Adeshina- DOI
- 10.2991/jaims.d.210512.001How to use a DOI?
- Keywords
- Deep learning; Genomics; DNA; Bioinformatics
- Abstract
The last few years have seen an advancement in genomic research in bioinformatics. With the introduction of high-throughput sequencing techniques, researchers now can analyze and produce a large amount of genomic datasets and this has aided the classification of genomic studies as a “big data” discipline. There is a need to develop a robust and powerful algorithm and deep learning methodologies can provide better performance accuracy than other computational methodologies. In this review, we captured the most frequently used deep learning architectures for the genomic domain. We outline the limitations of deep learning methodologies when dealing with genomic data and we conclude that advancement in deep learning methodologies will help rejuvenate genomic research and build a better architecture that will promote a genomic task.
- Copyright
- © 2021 The Authors. Published by Atlantis Press B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
The domain of bioinformatics has gained significance through the influence and relevance of Machine Learning (ML) methodologies. Many of the computational tools used in bioinformatics have been addressed by the ML community. Recent advancements in the Omics domain have brought about higher impactful collaborations between bioinformatics experts and ML experts. Several ML methodologies have proven to be very useful in solving bioinformatics related research questions, notably problems based on classification, clustering, and regression [1]. These methodologies are applicable to functional genomics, gene-phenotype association, gene structures, and gene interactions [2].
The emergence of “big data” has turned Deep Learning (DL) approaches into a discipline in ML. ML models are now considered to be effective and efficient to use to deal with big datasets [3] and have achieved a high prediction accuracy in real-life applications. However, they are still limited when compared with DL. The limitation of the ML methodologies lies in the fact that they are unable to handle raw data in their natural form [4]. Research has shown that the DL can provide models with higher accuracy [5] and, to a large extent, the models are efficient at discovering patterns which enables them to be applied in a range of domains including engineering, meteorology and medicine. Both the ML and DL require a training dataset. The training dataset in DL is more demanding and it affect the prediction value of the model.
DL models were first designed in the 1980s and were based on the concept of the Perceptron Model and the notion of neurons [5]. The models are at the heart of the predictive model for big datasets [6]. The need for huge computing power and large training datasets for DL models created a limitation for DL models until the introduction of high-performance graphics processing units (GPUs) with parallel architecture which makes computing more realistic. The motivation for this research is to understand the performance of DL architectures in bioinformatic tasks. In recent times, DL architectures have been applied in different fields, including natural language processing (NLP), computer vision, speech recognition, voice recognition and genomics analysis. An integral component of the DL models is the number of layers through which the data is transformed and this shows how “deep” the layer is in the design of the architecture. The DL network can have a multitude of layers, often hundreds, while the traditional neural networks are known for having only two or three layers. The choice of the DL network for the predictive process requires a great deal of parallelisms and special hardware for effective and sound prediction [7]. The DL models are known for hardware limitations and huge resource demands. To overcome these challenges, DL models have the capacity to scaleup the training phase when they use pipeline parallelism. Figure 1 shows some DL architectures. Figure 2 shows the benefits of using DL in bioinformatics in the discovery of splice junction from deoxyribonucleic acid (DNA) sequences, recognition of finger joints from X-ray images and detection of lapses from electroencephalography (EEG) [1].
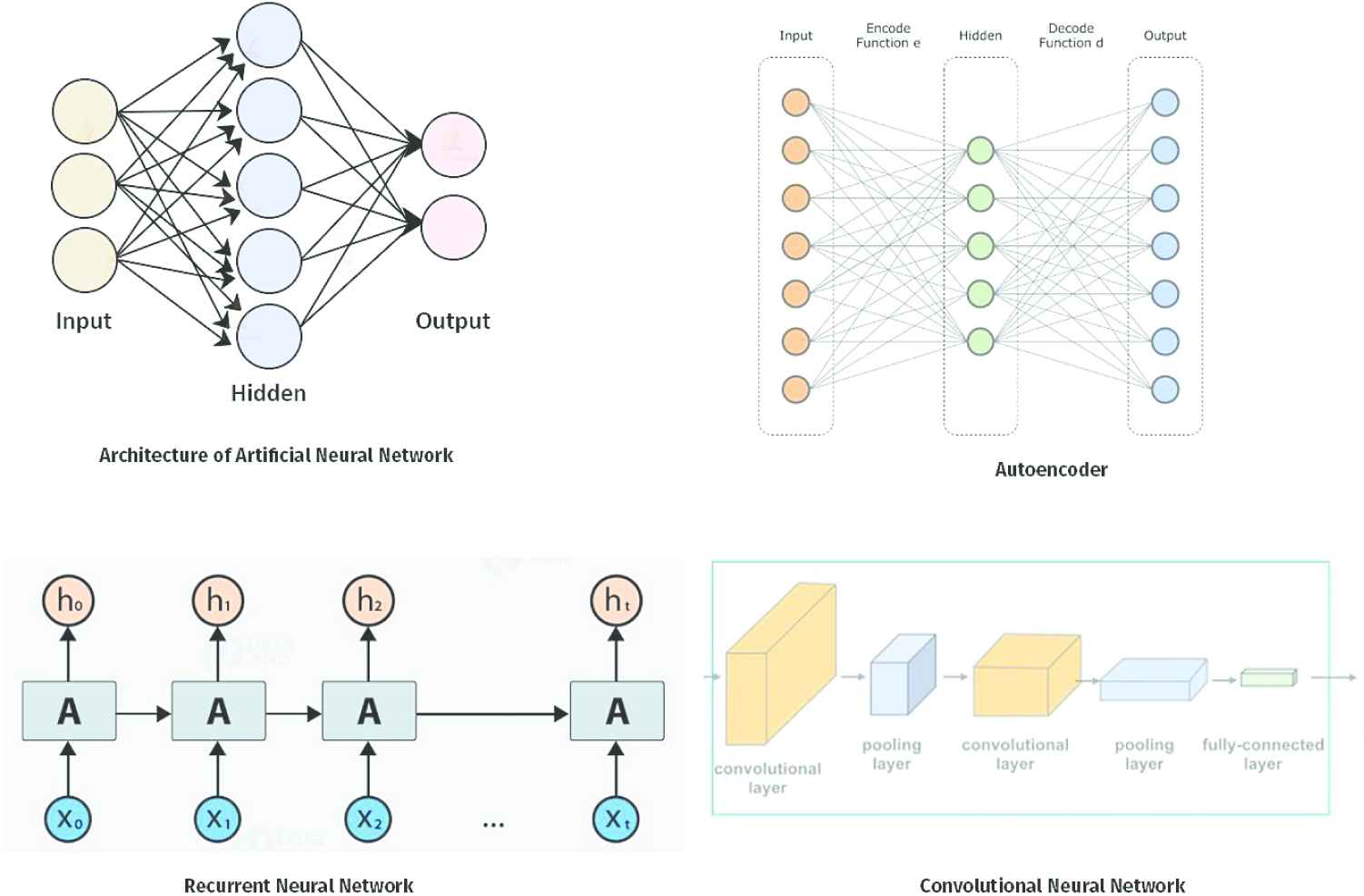
Showing deep learning architectures.
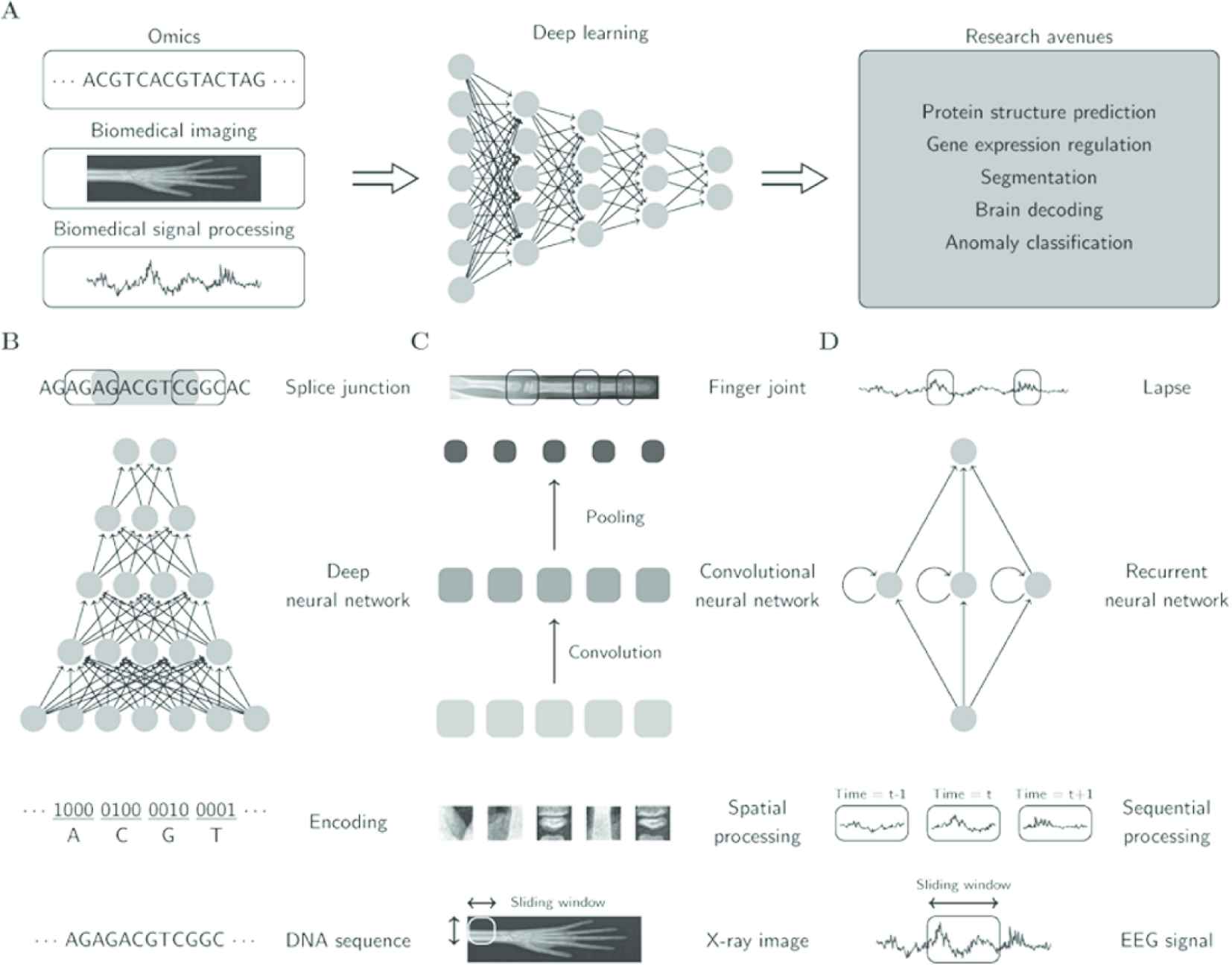
Deep learning models in bioinformatics.
The goal of genomic research is to understand the various genomes in different species. One notable highlight of genomic research is the ability to study how genetic factors interact with the surrounding environment under different conditions. The study of genomes involves understanding the genes that are possessed by an organism, while the study of genes is limited to the study of a specific number of genes. The study of the genomic makeup of homo sapiens involves searching through genetic components, encompassing 3 billion units of DNA, that contain ribonucleic acid (RNA) genes, long-rang regulating elements, protein-coding genes and transposable elements [8]. Advances in genomic research, for example, next-generation sequence technology that enables the entire DNA sequence of an organism to be readable, mean that this kind of research is becoming ever more data-intensive. With a high volume of facts and information generated by genomic research, there is huge potential for scientific-based research that incorporates statistical methods. The aim of using statistical methods is to identify the various genomic elements, such as introns, promoters, enhancers and exons. Models based on the DNA sequence can be built to provide insights into the biological mechanism of the genes. There are many data types that are readily available, such as genomic assays (RNA-seq expression), transcription factor (TF) binding chip-seq data and chromatin accessibility assays (DNase-seq, MNase-seq, FAIRE). A combination of different data can promote a better and deeper understanding of the genes. The majority of these datasets is available on most genomic portals such as NCBI (www.ncbi.nlm.nih.gov), GDC (www.portal.gdc.cancer.gov), Ensembl (www.ensembl.org).
The DL methodologies have helped provide high computation power to resolve complex research hypotheses in genomics [7]. Much has been written about how DL methodologies have helped to revolutionize the field of artificial intelligence. Advancements in DL architectures such as Convolutional neural network (CNN), recurrent neural network (RNN) and long short-term memory (LSTM) have helped push various architectural advancements in DL. The fusion of the DL methodologies and genomic research will promote a better understanding of genomics that will benefit many fields including medicine, pharmacy, agriculture, and so on. The field of medicine will transition from diagnostic gene therapies to personalized medicine when DL methodologies and genomic research is combined and it will bring about high-performance computing and a great abundance of genomic datasets. Therefore, the need to design powerful DL methodologies to enhance the development of the genomic industry necessary. This research is aimed at giving an in-depth overview of the most current DL application in genomic research.
This paper is organized as follows: Section 2 provides a description of DL architecture for genomic research. We describe the application of each DL architecture in solving the genomic problem. Section 3 describes the various advancements in DL architecture. Section 4 describes the resources and methodology used to identify the studies on DL architecture to be included in this review. The search process, the selection process, and the analysis of the selected studies were based on the research motivation. Section 5 provides an overview of the discussion around gene expression and gives an overview of gene transformation from DNA to mRNA and later to protein. Section 6 provides an overview of the discussion on the limitations of DL models in genomic research. Section 7 provides a general discussion about DL models. Finally, Section 8 concludes the paper and presents some possible future directions for further study.
2. DL ARCHITECTURE FOR GENOMIC
The different DL algorithms have their respective advantages in resolving specific problems in genomic applications. CNNs are known for capturing features in image classification tasks and have been adopted to automatically learn local and global characterization of genomic data [9]. RNNs are famous for speech recognition problems and are skilful enough at handling sequence data such as DNA. The autoencoders are known for denoising capacity and preprocessing of genomic data. At the point of designing DL models, the researcher could leverage the merits of DL algorithms to efficiently extract reliable features and reasonably model biological processes. This section reviews some aspects of some types of DL architectures, focussing on how researchers can leverage the advantages to benefit genomic research.
2.1. Artificial Neural Network
The most famous learning model is the artificial neural networks (ANNs) model that was introduced in the 1950s and is still actively studied to date [10]. Neural networks are made up of connected elements known as neurons, cells or nodes arranged into layers [11]. ANNs have an input layer where data enters the network, one or more hidden layers and one transformed output layer. Every hidden layer in the ANN is made up of several neurons, and each neuron is fully connected to all neurons in the previous layer. Each connection in the network is quantified by its weight. The weights need to be set to a favorable value, which is estimated through a training process for the network that is carried out to produce the correct output. Once the pattern is learned in the network, the network can be used to make predictions on new data, that is, to generalize to new data. ANNs are difficult and computationally expensive to train but they are flexible and are able to model and solve complicated problems [12]. Recently, AN Ns have become one of the prominent and most studied methods in ML. The growth in the use of ANNs is due to the growth of big data, the availability of powerful processors for parallel computations, the ability to tweak the algorithms used in constructing and training the networks, and the development of frameworks that are easy to use.
2.2. Convolutional Neural Network
CNNs are known to be one of the most successful DLmodels for image processing because of their ability to analyze spatial information. The early adoption of CNNs in genomics was in the area of computer vision [13]. The adaptation of CNNs from computer vision to genomics was made possible by assimilating a window of genome sequencing as an image. Research in this area represented the genome sequence as a fixed length of a 1D sequence window with four channels (A, C, G, T) rather than 2D images with three color channels (R, G, B). With this, CNNs can perform a single sequence through the 1D convolutional kernel. The important feature of CNNs is their ability to perform adaptive feature extraction during the training process. For example, CNN can determine meaningful recurring patterns with small variances, such as genomic sequence motifs [14].
CNNs have outperformed other existing methods in the domain of sequence-based problems. CNNs were successfully applied to model sequence specificity of protein binding [15,16]. A convolutional three-layer network of CNN models was developed to predict the effects of noncoding variants of TF binding, DNA accessibility and histone marks of sequences from the only genomic sequence. Research has shown that CNN has surpassed other existing methods [15], so developing structures that are not appropriate would yield a poorer result than convolutional models. The ability to match a CNN architecture to a given task lies in harnessing the power of CNNs. Researchers are expected to have a better and more detailed understanding of CNN architectures and also take into account biological knowledge. A parameterized CNN was developed to conduct a systematic exploration of CNN on two classification tasks: motif discovery and motif occupancy. They performed a hyperparameter search using Mri 6 and examined the performance of nine variants of CNNs. The researchers concluded that CNNs do not necessarily have to be deep to carry out the motif discovery task as long as the structure is appropriately designed. In genomic research, since DL models are always over-parameterized, simply changing the network depth would not account for much improvement in model performance. Researchers should pay more attention to particular techniques that can be used in CNNs, such as the kernel size, the number of the feature map, the design of pooling or convolution kernels and the choice of window size of input DNA sequences [16].
2.3. Recurrent Neural Network
There has been a surge of interest in RNNs following impressive results obtained in challenging sequential prediction problems such as NLP, language translation and speech recognition. RNNs outperform CNNs and other deep neural networks on data that is highly dependent on the ordering of the sequence in memorizing long-range information through loops in networks. The input data are processed sequentially by RNNs. Past information can be stored implicitly by recurrent computation in the hidden state units where cyclic connections exist, then the model output will be an integrated result considering the current input and all previous inputs. Bidirectional recurrent neural networks (BRNNs) were proposed for other scenarios where both past and future inputs matter [4]. The cyclic structure makes a seemingly shallow RNN over long-time prediction very deep if unrolled in time. To resolve the problem of vanishing gradient created by this fact, the hidden unit of the RNNs is substituted with LSTM units to truncate the gradient propagation appropriately [17]. RNNs have several applications in bioinformatics. They are used for genome base calling [18], quantification of noncoding DNA [19] and protein prediction of subcellular location from protein sequences [20].
2.4. Autoencoder
An autoencoder is a form of ANN that is known for learning efficient data coding in an unsupervised manner. Autoencoders are used as preprocessing tools to initialize the network weight but, in recent times, the range of autoencoders has been extended to include stacked autoencoders (SDAa), denoising autoencoders and contractive autoencoders among others [8]. Autoencoders have recorded many success stories relating to the task of feature extraction because they can learn a compact representation of input through a procedure known as the encode–decode procedure [8]. Many autoencoder variants have been applied in different applications. For example, a SDAs has been applied for gene clustering tasks [21]. Autoencoders have also been used for the task of dimensionality reduction in gene expression [22]. A very important fact to note about the application of autoencoders is that a better reconstruction accuracy does not necessarily translate into model improvement [23]. Table 1 below shows the purpose and strength of the DL architectures discussed in this section.
| Paper | Purpose | Strength |
|---|---|---|
| [10] | To investigate the historical background of ANNs and their applications within the healthcare system. | The researchers identified the strength of ANN and other forms of deep learning model applications in healthcare. |
| [12] | To capture an extensive and comprehensive discussion about ANNs and other machine learning models. | A complete chapter was dedicated to ANNs. The various transformation stages of ANNs over the years from a historical point of view were all captured in the chapter. The chapter also captured the mathematical building blocks of ANNs based on supervised learning and unsupervised learning. |
| [13] | To develop a deep learning model (CNNs) to classify the images in the ImageNet 2010 contest. | On the dataset, the authors achieved a top-1 and top-5 rate of 37.5% and 17.0% in 2010. In 2012, the authors achieved a top-5 test error rate of 15.3% using CNNs compared to 26.2% that was achieved by the second-best entry. |
| [14] | To develop a deep coevolutionary network to classify genomic sequence on transcription factor binding site task. | The system developed by the authors is known as Deep Motif (DeMo). The DeMo was able to extract motifs that are similar to and, in some cases, outperformed the current well-known motifs. The research also shows that a deep model consisting of several coevolutionary layers can outperform a single convolutional and fully connected layer. |
| [15] | To show how prostate segmentation in TRUS images informed by MRI priors can improve prostate segmentation that relies only on TRUS images. | The authors proposed a TRUS segmentation technique that is fully automatic and uses MRI priors. The algorithm used a convolutional neural network to segment the prostate in TRUS images. The methodology achieved more accurate segmentation of the base and apex with MRI segmentation. |
| [16] | To harness the power of CNN architecture for computational biology application. | The authors presented a systematic exploration of CNN architectures for predicting DNA sequence binding using a large compendium of transcription factors dataset. The best performing architecture was achieved by varying CNN width, depth, and pooling designs. The research showed that additional convolutional kernels to a network are important for the motif-based task. |
| [8] | To capture the strength of different deep learning models from a genomics perspective. | The research was able to present a concise view of autoencoder deep learning applications in various aspects of genomic research. |
| [21] | To demonstrate the use of an autoencoder as a preprocessing step for a popular learning task. | The autoencoder architecture was used to regenerate gene expression time-series data for two different datasets. The autoencoder’s performance was promising when tested with two popular datasets. |
| [22] | To develop computational methods that will facilitate hypothesis generation and biological interpretation of genomic datasets. | The authors developed a methodology known as Analysis using Denoising Autoencoder of Gene Expression (ADAGE). ADAGE was able to identify differences between strains, model the cellular response of low oxygen and predict the involvement of biological processes based on low-level gene expression differences. |
| [23] | To use a variational autoencoder to improve the accuracy of drug response prediction. | The authors developed the Drug Response Variational Autoencoder (Dr.VAE) model. The model outperforms the current Benchmark between 3 to 11% AUROC and 2 to 30% AUPR. It was noted in the research that better reconstruction accuracy does not necessarily translate into improved classification accuracy. |
| [4] | To describe deep learning techniques used by practitioners in industry, including deep feedforward networks, sequence modellng and practical methodology. | The research captures a wide range of discussion in relation to deep learning such as probability and information theory, numerical computation and machine learning. |
| [17] | To investigate the feasibility of using adversarial training for a sequential model (RNNs) with continuous data and evaluate the model using classical music in freely available midi files. | The results of the experiments show that adversarial training helps the model learn patterns with more variability and a larger tone span. The ability of the model to output more than one tone per LSTM call helps to generate music with a higher polyphone score. |
| [18] | To come up with an open source DNA base caller using a deep recurrent neural network. | The authors employed a carefully crafted RNN to show that availability of an open source tool with high base scaling accuracy will be very useful for the development of new applications. |
| [19] | To develop a predictive model of noncoding DNA. | The researchers proposed DanQ, a novel hybrid convolutional and bidirectional long short-term memory recurrent neural network framework for predicting noncoding functions from sequence. DanQ improves considerably upon other models across several metrics. |
| [20] | To demonstrate that LSTM networks can predict the subcellular location of proteins. | The authors showed that the LSTM model can predict the subcellular location of protein given only the protein sequence. Accuracy of 0.902 was achieved which surpasses other state-of-the-art algorithms. |
Deep learning architecture purpose and strength.
3. UPCOMING ARCHITECTURES
DL models have constantly shown some level of success in genomics. The expectation of researchers in bioinformatics from DL models is higher accuracy. This goes beyond outperforming statistical or ML methods. The most recent work on genomic problems is an approach beyond classical DL architecture to more advanced models. In this section, we review some emergent DL architectures that are skilfully modified or a combination of some classical DL models.
3.1. Enhancement of Classical Models
New architectures emerge from modifications made to the classical DL models. Researchers use their intuition to solve genomic problems by designing a suitable model. The work of [24] was motivated by the fact that protein folding is known to be a progressive refinement rather than an instantaneous process. The deep spatio-temporal neural network (DST-NNs) architecture was designed for the residue to residue contact prediction. The architecture consists of 3D stack neural networks that have the same topological structure (same input, hidden and output layer sizes) for each stack. Every stack level in the network is regarded as a distinct contact predictor and can be trained in a supervised manner to refine the predictions provided in the previous level. The refinement helps solve the problem of vanishing gradient in deep architectures. The spatial feature of the DST-NNs architecture references the original model inputs, while the temporal features are altered from time to time to progress the upper layers. The DeepCpG (deep cytosine and guanine separated by only one phosphate group) [25] is a deep architecture for predicting the methylation state of CpG dinucleotide in multiple cells. The DeepCpG consists of two CNNs and pooling layers to identify predictive motifs from the local sequence context and one fully connected layer to model motif interactions. The DeepCpG architecture allows the input of incomplete DNA methylation profiles to discover the predictive sequence motifs and also to quantify the effect of the sequence mutation [25]. The CpG model scans the neighborhood of multiple cells row by row, using a bidirectional gated recurrent network (GPU). The compressed features are yielded in a vector of constant size. The higher-level feature derived from the DNA-and-CpG model to predict methylation states in the cells is learned by the interaction of the joint model.
3.2. Deep Hybrid Architecture
The strengths of every DL architecture inspire researchers to develop a hybrid architecture that could leverage the potentials of multiple DL architectures. A hybrid convolutional and recurrence deep neural network (DeepQ) to predict the function of noncoding DNA from the sequence was developed by [19]. The input to the DeepQ architecture is a DNA sequence that is represented as a one-hot representation of the four bases. This input goes into a CNN with the aim of scanning motif sites. Motifs are known to follow a regulatory grammar and are governed by the physical constraint that is involved with their spatial arrangements and frequencies of combinations of DNA sequences. The motifs learned by CNN are then fed into the bidirectional long short-term memory (BLSTM) [19]. The Deep GDashboard was developed by [14]. The Deep GDashboard is a suite of visualization strategies to extract motifs or sequence patterns from the deep neural network model for transcription factor binding site (TFBS) classification. The understanding of the Deep GDashboard was demonstrated with three deep architectures: convolutional, recurrent and convolutional-recurrent networks (CNN-RNN), and the features generated by each network were validated through visualization techniques. The experimental results of the TFBS classification task show that the CNN-RNN outperformed the CNN or RNN alone. The visualization based on features achieved by Deep GDashboard shows that CNN-RNN architecture can model both motifs as well as dependencies among them [14].
4. RESOURCES AND METHODOLOGIES
This study aims to provide a concise review of DL methodologies within the genomic domain. The study did not capture studies that focus on radio-genomics DL architecture that are used for image capturing purposes [26]. The study focuses on literature that involves DL models being applied in gene expression. For the literature review, the narrative and scoping literature review approach was adopted [27] and a research search strategy was developed. This is illustrated in Figure 3 below.
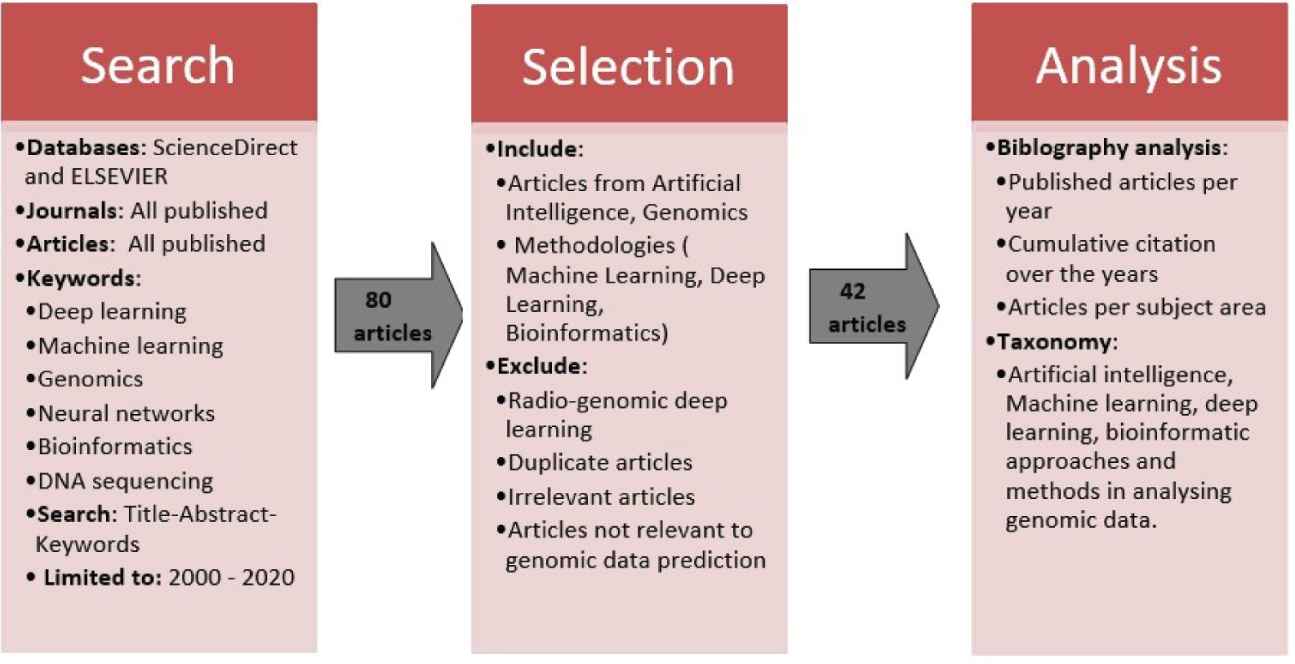
Process flow of the search, selection and analysis of the comprehensive literature review.
The RNN and LSTM were used by DeepTarget [28] and deep MirGene [29] respectively for micro ribonucleic acid (miRNA) and target prediction using expression data. Both the DeepTarget and deepMirGene algorithms proved that miRNA can be predicted more accurately than when using a non-DL model such as TargetScan [30]. The DL model does not require any handcrafting features that are used in the other non-DL model. Tables 2 and 3 show the application of RNN and LSTM DL models in genomics.
| Name | Publication | Omic Dataset | Purpose | Accuracy | Performance Gap |
|---|---|---|---|---|---|
| DeepTarget | [28] | Size | prediction | 0.96 | +25% F-measure Outperformlinear |
| D-GEX | [33] | Expression of Landmark genes | Gene expression inference | An overall error of 0.3204 ± 0.0879 | regression and KNN in most of the target genes |
List of works showing the application of RNN deep learning model in genomics.
| Name | Publication | Omic Dataset | Purpose | Accuracy | Performance Gap |
|---|---|---|---|---|---|
| DeepMirGene | [23] | Positive premiRNA and non-miRNA | miRNA target | 0.89 Sensitivity | +4% f-measure |
| AttentiveChome | [29] | Histone modification | Classify gene expression | AUC = 0.81 | Marginally better than DeepChrome |
List of Works showing the application of LSTM deep learning model in genomics.
The first approximation on how best to apply a multi-layer free-forward ANN to analyze RNA-seq gene expression data was presented in [31]. The free-forward ANN model outperformed the Least Absolute Shrinkage and Selection Operator (also known as LASSO) in analyzing RNA-seq gene expression profiles data. An effective approach for using a deep network as a preprocessing step for clustering gene expression data was demonstrated by Gupta [21]. The authors used a DL model in the preprocessing step that involved clustering the yeast expression microarrays into modules that simulate the cell cycle processes. The final result showed that the DL methodology outperformed the principal component analyses (PCA) algorithm. To achieve a better result, the authors used deep aelief with auto encoder (AE) for the learning process which is an unsupervised learning approach for gene selection. In the recovery of organzsation transcriptomic machinery [32] used AE on yeast complementary seoxyribonucleic acid (cDNA) microarray data to learn the encoding system of yeast transcriptomic machinery.
A special case of AE is the shallow denoising AE in which the model feeds the input data with noise. This special AE has been evaluated for its usefulness in the domain of genomics. An analysis of auto encoders of gene expression (ADAGE) was carried out by [22] on publicly available gene expression data to identify differences between strains and predict the involvement of biological processes based on low-level gene expression differences. In term of gene data expression inferencing, the authors of deep learning for gene expression (D-GEX) provided a deep architecture to infer the expression of target genes from the expression available on landmark genes [33]. The sum of 111,000 public expression profiles from gene expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) was used by D-GEX which trained a multi-layer feedforward deep neural network with three hidden layers. The results showed that the DL model provided better accuracy than the linear regression when it came to inferring the expression of human genes (about 21,000) based on the landmark genes (about 1,000). Although the DL model achieves a higher level of accuracy when compared with other existing ML models, the architecture of the model still needs to be improved as its performance is still quite poor. Tables 4 and 5 show the application of autoencoder and ANN DL models in genomics.
| Name | Publication | Omic Dataset | Purpose | Accuracy | Performance Gap |
|---|---|---|---|---|---|
| DeepNet | [18] | Time-series gene expression | Preprocessing steps for clustering | - | Better than PCA |
| ADAGE | [19] | cDNA Microarrays | Organization of transcriptomic prediction | - | Significant overlap with previous studies |
| DeePathology | [34] | mRNA and miRNA | Predicts tissue of origin, normal or disease state and cancer types | 99.4% accuracy for cancer subtypes | 95.1% for SVM |
List of Works showing the application of Autoencoders based deep learning model in genomics.
| Name | Publication | Omic Dataset | Purpose | Accuracy | Performance Gap |
|---|---|---|---|---|---|
| DeepNet | [25] | RNA-seq | Control cases | 0.7 | Same of worst AUC from LASSO |
| DeepVariant | [32] | Cell-line with drug response | Predict drug response | AUC = 0.65 | Outperform RF 0.54 AUC |
List of Works showing the application of ANN deep learning model in genomics.
A CNN method known as DeepChrome was used to automatically learn combinatorial interactions among histone modification marks for gene expression prediction purposes [34]. DeepChrome accuracy prediction outperforms other existing models, such as support vector machine and Random Forest for Boolean (high/flow) gene expression using histone modification as input. AttentiveChrome [35] is another variant of DeepChrome that was developed by the same authors of DeepChrome. The AttentiveChrome is an LSTM model developed to enhance the capacity of the DeepChrome using a unified architecture to interpret dependencies among chromatin factors to control gene regulation. Another CNN architecture is the Deep Variant [36]. This is a CNN caller that proved to outperform all non-DL state-of-the-art variant callers. Using different versions of the human genome built for training and testing, the deep variant presents a generalization that goes beyond just the training dataset. The deep variant was trained on an independent set of samples and was tested against a mouse dataset; it achieved a level of accuracy that outperformed training on the mouse data itself. To predict locus-specific signals from epigenetic assays using a DNA sequence, the DeepFIGV DL model was used [37]. The DeepFIGV can model quantitative variation in the epigenome using many experiments from the same cell type and assay by integrating the whole-genome sequencing to create a personalized genome sequence for each individual.
A CNN model for predicting response to therapy in cancer was implemented in [38]. The training model’s task was to predict drug response using a pharmacogenomic database of 1001 cancer cells. The DL method outperformed the current state-of-the-art ML frameworks for this specific task. Table 6 show the application of CNN DL models in genomics. A multimodal deep belief framework that can integrate DNA methylation, miRNA, and gene expression data for the identification of cancer subtypes was proposed by [39]. The proposed method exploits the complex cross-modality correlation and the deep intrinsic statistical properties among multi-platform input data.
| Name | Publication | Omic Dataset | Purpose | Accuracy | Performance Gap |
|---|---|---|---|---|---|
| DeepChome | [28] | Histine modification | Classify gene expression | 80% AUC | +5% from SVM +2% from RF |
| DeepVariant | [30] | Whole-genome sequence | Variant caller | 99.45% FI | Produce more accurate results |
| DeepFIGV | [31] | Whole-genome sequence | Predict quantitative epigenetic variation | z-scores DNase rho = 0.0802, P = 5.32e-16 |
List of Works showing the application of CNN deep learning model in genomics.
The DeePathology is another multimodal DL model in genomics. The DeePathology is a DL method used in the area of gene inferencing. The method can simultaneously infer various properties of biological samples, through multi-task and transfer learning. The model can accurately predict tissue and disease type and it does this by encoding the whole transcription profile [40].
5. EXPRESSING GENE
Gene expression is the process of converting the genetic instructions in the DNA into functional products such as proteins and other molecules. The gene encodes proteins and proteins dictate cell functions. The genetic code of the gene, which is also known as the nucleotide sequence, is used to regulate the cells’ function and direct protein synthesis. The self-regulation of the cells is achieved by adjusting the amount and type of proteins the nucleotide produces. The gene expression process allows for cells to respond to the ever-changing environment and help the cells to self-regulate by adjusting the amount of proteins the gene produces. A review of some research studies that applied DL to analyze how the gene is expressed and regulated is presented in the next sub-section. Figure 4 shows how a pair of DNA strands is expressed to proteins.
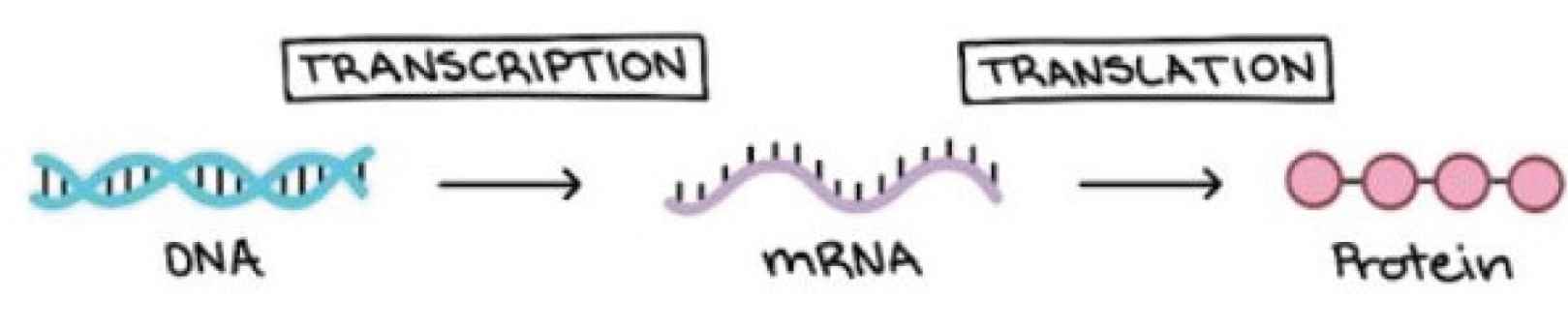
Show how gene is expressed from a pair of DNA strand to proteins.
5.1. Characterization of Gene Expression
Historically, gene expression is measured by low-throughput fluorescence-based methods, microarray technologies, quantitative polymerase chain reaction (qPCR), and so on, and in recent time changed to performing RNA sequencing (RNA-seq) to catalogue whole transcriptomes. Gene expression profiling, which is the process of measuring the activity of thousands of genes at once, has been used to determine the cellular state in response to genetic perturbations, drug treatment and various diseases. There has recently been a breakthrough in gene expression profiling and this has helped to lower the costs of whole-genome gene expression. However, the costs of engaging an academic laboratory to perform a whole-genome gene expression profile over a large number of conditions remain very high. There has been an increase in the number of genome-wide gene expression assays and these cut across different species and are publicly available in the form of a database. For example, the connectivity Map (cMap) project that maps molecules that are functionally connected was created to serve as a reference point for collection of gene expression profiles [35]. The database facilitates the computational model for the biological interpretation of these data. The application of principal component analysis (PCA) on gene expression data to capture the various gene clusters has shown that the PCA tool has failed to capture some biological considerations, rendering it ineffective [41]. In 2014, Tan and others presented an unsupervised features construction and knowledge extraction approach to genome-wide assay. The approach helped capture the key biological principles in breast cancer with the application of a stacked denoising autoencoder [42]. The ADAGE project was used to extract relevant patterns from the large-scale gene expression datasets. An enhanced ADAGE was presented in 2016 by the same authors. The improved ADAGE can construct features that contain both clinical and molecular information [22]. The authors were able to uncover similarities among genes that share the Kyoto Encyclopedia of Gene and Genomes (KEGG) pathways without prior knowledge. In 2017, Tan and others also developed an ensemble ADAGE (eADAGE) to integrate stable signatures across models [2]. The three studies are very similar and all used the Pseudomonas aeruginosa gene expression datasets. In 2015, Gupta demonstrated the efficacy of using enhanced data by using a multi-layer denoising autoencoder. The multi-layer denoising autoencoder was used to cluster yeast expression microarrays into known modules representing cell cycle processes [21]. In 2016, Chen was motivated by the hierarchical organization of yeast transcriptomic machinery. He used a four-layered autoencoder network with each layer accounting for a specific biological process in the gene expression. This research introduced sparsity into the autoencoders [32]. The work demonstrated denoising autoencoders over PCA and independent component analysis (ICA). The unsupervised model helped identify gene signatures that may have been overloaded.
6. LIMITATIONS OF DL MODEL IN GENOMIC
The application of the DL model in genomic mapping is in the early stages and a great deal of work still needs to be done given the limitations that are associated with applying DL methodologies to Omic datasets. The following are some of the limitations:
6.1. The Curse of Dimensionality
This is the most pronounced limitation in artificial intelligence when Omic datasets are applied with DL models [43]. Genomic datasets are known for presenting a huge number of variables and a small number of samples. The genomic domain is considered to be a big data domain in terms of the data volume. The fact that Omic datasets have a smaller number of samples presents a huge problem for genomic, DL, and ML algorithms [44]. Although some repositories provide access to public genomic datasets, a great deal of preprocessing and harmonization has to be performed on these public datasets for DL training tasks.
6.2. Class Imbalance
It is a known fact that genomic datasets that are sourced from various public repositories usually feature an inherent class imbalance and DL models cannot be effective until sufficient instances per class are available. Research has shown that transfer learning can help deal with the challenge of the imbalance problem [42] since the model can be trained with a general dataset [45].
6.3. Heterogeneity of Data
The genomic dataset is heterogeneous. Genomic data includes (i) sequenced genes; (ii) genome alternation; (iii) gene expression profiles; (iv) gene interaction at a biological systems level; and (v) gene variants. One of the limitations of integrating this subgroup is the interdependencies among these heterogeneous groups.
6.4. Interpretation of Model
Model interpretation is one of the issues for DL architecture [39]. In the bioinformatics domain, researchers prefer the white box approach over the black box approach because it is very difficult to understand the learned pattern and extract the relationship between the data and outcomes due to the structure of DL models.
6.5. Parameter and Hyperparameter Tunning
Model tunning is one of the difficult steps in DL. Analyzing the initial results helps the tunning process because parameter tuning correlates with research questions and datasets. The hyperparameter tuning for DL architectures is the learning rate, batch size, momentum, and weight decay. If any of the hyperparameters is wrongly tuned it may result in underfitting or overfitting of theDL models [46].
7. FUTURE PERSPECTIVE
There is a rapid evolution in the methods for heterogeneous data and source integration in bioinformatics and computational biology. The need to improve on the ability to describe and represent biomedical findings on different omics datasets is very important. Neural networks have been adopted since the 1950s in solving problems. Some level of progress has been recorded in recent times in DL methods due to a better understanding of the learning systems, decline in computational cost, integration of different technologies and increase in computational power. However, the limitation of DL models discussed in the previous section are still situations where the model fails, underperform and need to be improved. DL models have an advantage over other genomics algorithms in the preprocessing steps that traditionally are known to be error-prone and time-consuming. DL methods require huge computing power and memory, thus they are not required to be applied to moderate-size genomic datasets. A better understanding of DL methods will provide a better interpretation of the model. To mitigate the shortcomings of DL methods, there is a need to improve the existing theoretical foundation of DL based on experimental data, this will help the performance of individual neural network model.
8. CONCLUSION
The genomic domain is a very challenging area for the DL model when compared to other domains such as computer vision, speech recognition, and NLP. This is because of our inability to interpret genomic information. Based on the studies reviewed above, it is obvious that the rejuvenation of DL methodologies can help promote better state-of-the-art architecture in the genomic domain that will solve the genomic task. DL methodologies have led to better results in genomics than some computational methods with regards to predictive performance, although they lag behind traditional statistical inference methods when it comes to interpretation. The predictive performance of DL methodologies has not reached real-world application expectations. There is the need to make a conscious effort to analyze and compare datasets that are privately and publicly available together to increase the role of DL methodologies in genomic prediction and prognosis.
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
AUTHORS' CONTRIBUTIONS
Y. A. Abass wrote the manuscript with contributions from S. Adeshina.
ACKNOWLEDGMENTS
The authors thank the National Information Technology Development Agency (NITDA) and Nile University of Nigeria (NUN) for supporting Y.A. Abass studies in Nigeria. The authors thank the anonymous reviewers for their objective remarks, and for their suggestions on the deep learning architectures discussion, which greatly contributed to this paper.
REFERENCES
Cite this article
TY - JOUR AU - Yusuf Aleshinloye Abass AU - Steve A. Adeshina PY - 2021 DA - 2021/05/19 TI - Deep Learning Methodologies for Genomic Data Prediction: Review JO - Journal of Artificial Intelligence for Medical Sciences SP - 1 EP - 11 VL - 2 IS - 1-2 SN - 2666-1470 UR - https://doi.org/10.2991/jaims.d.210512.001 DO - 10.2991/jaims.d.210512.001 ID - Abass2021 ER -