
Passive Immunity Should and Will Work for COVID-19 for Some Patients

- DOI
- 10.2991/chi.k.210328.001How to use a DOI?
- Keywords
- COVID-19; coronavirus; plasma; antibody; immunity
- Abstract
In the absence of effective antiviral chemotherapy and still in the context of emerging vaccines for severe acute respiratory syndrome-CoV-2 infections, passive immunotherapy remains a key treatment and possible prevention strategy. What might initially be conceived as a simplified donor–recipient process, the intricacies of donor plasma, IV immunoglobulins, and monoclonal antibody modality applications are becoming more apparent. Key targets of such treatment have largely focused on virus neutralization and the specific viral components of the attachment Spike protein and its constituents (e.g., receptor binding domain, N-terminal domain). The cumulative laboratory and clinical experience suggests that beneficial protective and treatment outcomes are possible. Both a dose- and a time-dependency emerge. Lesser understood are the concepts of bioavailability and distribution. Apart from direct antigen binding from protective immunoglobulins, antibody effector functions have potential roles in outcome. In attempting to mimic the natural but variable response to infection or vaccination, a strong functional polyclonal approach attracts the potential benefits of attacking antigen diversity, high antibody avidity, antibody persistence, and protection against escape viral mutation. The availability and ease of administration for any passive immunotherapy product must be considered in the current climate of need. There is never a perfect product, but yet there is considerable room for improving patient outcomes. Given the variability of human genetics, immunity, and disease, and given the nuances of the virus and its potential for change, passive immunotherapy can be developed that will be effective for some but not all patients. An understanding of such patient variability and limitations is just as important as the understanding of the direct interactions between immunotherapy and virus.
- Copyright
- © 2021 International Academy for Clinical Hematology. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
Interest in passive immune therapy for the treatment of infectious diseases has long existed [1–3]. There have been many examples of application for either prevention or active treatment, but variable efficacies occur in the many scenarios in which such treatment has been applied. During the epidemic of severe acute respiratory syndrome (SARS), equivalent approaches were resurrected, but largely thereafter abandoned, as the SARS prevalence came to a conclusive end in a relatively short period of time. Contemporary but meagre progress had been made in response to the Middle East Respiratory Syndrome (MERS), but the COVID-19 pandemic has again rekindled considerable interest. Indeed, a recent large randomized placebo-controlled trial of convalescent plasma infusion was conducted in which there was a lack of efficacy for severely ill patients on either clinical status or mortality; that analysis has reminded the medical and scientific communities to approach any such therapy with due diligence [4]. The lack of obvious effective antiviral therapy continues to be a source of frustration despite the advent of several seemingly effective vaccines. As vaccine distribution widens and long-term vaccine efficacy continues to be gauged, active treatment of COVID-19 is yet in need of other preventive or treatment strategies. In the context of the publication of Simonovich et al. [4] and its implications to passive therapy for SARS-CoV-2 infections, this review examines the promise, application, and future of passive immunotherapy.
2. LESSONS FROM COMPARATIVE CORONAVIROLOGY
Preceding or shortly following SARS and apart from MERS, and COVID-19, several coronaviruses were found to cause human respiratory illnesses and less common complications [5]. None of these four virus (OC43, 229E, NL63, HKU1) infections were effectively studied for passive immunotherapy or vaccination, but it was shown at least for some that serum neutralizing or respiratory antibody presence correlated with protection [6]. Similar themes on protection were also evident through experimentation or natural infection with animal respiratory or enteric coronaviruses [7]. For the latter, passive immunotherapy proved effective, and this was especially typified by the understanding of protective antibody transfer from mother to offspring [7]. Table 1 highlights some key findings in passive immunity for these coronaviruses which have relevance to similar efforts for SARS-CoV-2 infections [8,9]. Passive immunity has potential for protection and treatment, but there is usually a dose- and time-dependent effect. This is applicable to convalescent plasma, hyperimmune serum, purified immunoglobulin, or monoclonal antibodies. Different monoclonal antibodies behave variably but are more often better active in combinations.
| References | Model system | Treatment | Outcomes |
|---|---|---|---|
| Human endemic respiratory coronavirus infections | |||
| [8] | Human experimental infection with 229E | None; volunteers examined for preceding immunity | Viral challenge modulated by previous immunity; both circulating and local antibody were associated with protection |
| [9] | Human experimental infection with 229E | None; repeat infections after 1 year | Intranasal infection protects against repeat nasal challenge; post-infection antibody wanes; those primarily challenged but not seemingly infected maintained secondary challenge reduction in illness |
| SARS-CoV-1 infection | |||
| [10] | Murine | Post-infectious serum given intraperitoneal; equivalent to 700–1750 mL/70 kg person | Reduced lung titres of virus post-challenge; dose-responsiveness |
| [11] | Murine | Neutralizing human MAb given intraperitoneal; 4–80 mg/kg | Reduced lung titres of virus post-challenge and reduced histopathology of disease |
| [12] | Murine | Engineered human MAb given intraperitoneal; 200 mcg | Prevented and mitigated infection |
| [13] | Human | Convalescent plasma 1:160–1:2560 ‘antibody titre’ given in dose of 200–400 mL intravenous | Reduced hospital stay and mortality (note: method of antibody assessment not mentioned) |
| [14] | Human | Convalescent plasma 1:160–1:2560 ‘antibody titre’ given in dose of 160–640 mL intravenous 9–22 days | Trend for reduced mortality and reduction in some disease parameters; timing-dependent (note: method of antibody assessment not mentioned) |
| MERS infection | |||
| [15] | Murine | Humanized murine MAb given intravenous 10 mg/kg | Reduced lung titres of virus and lung disease |
| [16] | Murine | Humanized murine MAb given intravenous 2 mg/kg | Reduced lung disease and mortality |
| [17] | Murine | Human MAb given intraperitoneal; 1–200 mcg | Reduced lung titres or virus and lung disease two MAb were superior to any one |
| [18] | Murine | Chicken IgY given intraperitoneal 500 mcg twice | Given after onset of infection and improved some infection parameters |
| [19] | Murine | Equine serum (200 mcL), equine IgG (500 mcg), or F(ab′)2 (500 mcg) given intraperitoneal | Reduced lung titres of virus |
| [20] | Murine | Human convalescent serum (100 mcL titre >1:5000) or human MAb (20 mcg) given intraperitoneal four times | Reduced viral load in lungs and improved survival |
| [21] | Marmoset | Human MAb given intravenously; 10–25 mg/kg | Effected prevention more than treatment of active infection |
| [22] | Marmoset | Hyperimmune marmoset plasma (1 mL; titre 1:3840) or single human MAb (5 mg) given intravenously early after infection and repeated subcutaneously later | Both decreased signs of disease but only plasma reduced viral load |
MAb, monoclonal antibody.
Key translational findings in studies of passive immunity for human endemic respiratory coronavirus, SARS-CoV-1, and MERS infections
There has been considerable debate over the role of human endemic respiratory coronaviruses for providing some protection for SARS-CoV-2 infections [23–27]. The finding of common and conserved epitopes initially provided the stimulus, but a more precise analysis of antibody evolution and cell-mediated immune reactivity led to further insight. Neutralizing cross-reactive antibodies between these viruses and SARS-CoV-2 are generally lacking in humans, and yet there are some pan-coronavirus cross-reactive epitopes [26–28]. Dugas et al. [29] have found evidence suggesting a lesser severe disease with COVID-19 if patients possessed higher levels of antibodies to the endemic coronaviruses. Others found a rise in antibody levels to endemic respiratory coronaviruses during SARS-CoV-2 infection [30]. Also, immunity from other coronavirus infections might protect against COVID-19 over time, and susceptibility to serious infection appears to be age-accrued. These issues may be explained in part by differences in T cell responses and background humoral immunity [31].
3. COVID-19
3.1. Immunogens and Humoral Immunity
Characterization of viral immunoreactive epitopes is becoming more apparent as virus-directed antigens are being better characterized [32]. The tools for any such characterization were largely available prior to the onset of the pandemic, and further progress often relies on the application of past knowledge to the context of SARS-CoV-2.
The Spike (S) protein is a dominant immunoreactive protein and has several recognized regions, including the receptor binding domain [33–39]. Other recognized antigens commonly include Nucleocapsid (N), Membrane (M), orf3a, orf6, orf8, orf10, and Nsp5 (Mpro or 3CLpro) (Figure 1) [36,37,39–44]. The latter may or may not include structural proteins of intact virus and may arise as proteins derived during the processes of infection. There are potentially several more virus-directed proteins that may attract humoral responses [40]. Some of the latter appear to be shared with common non-SARS-CoV-2 coronaviruses [34,45]. For unknown reasons, the Envelope (E) protein which is virus surface-exposed does not attract humoral immune reactivity very well [46]. Antigen-specific antibody responses can vary, and stereotypic de novo responses occur [34,35,37,47–49]. An immunodominance of anti-Receptor Binding Domain (RBD) in the neutralizing antibody profile has been proposed [33,50]. Others found a lesser dominance in anti-RBD [34,48]. Smits et al. [44] suggest that the N protein is immunodominant. Recognition of immunogenic proteins or other structures is yet provisional.
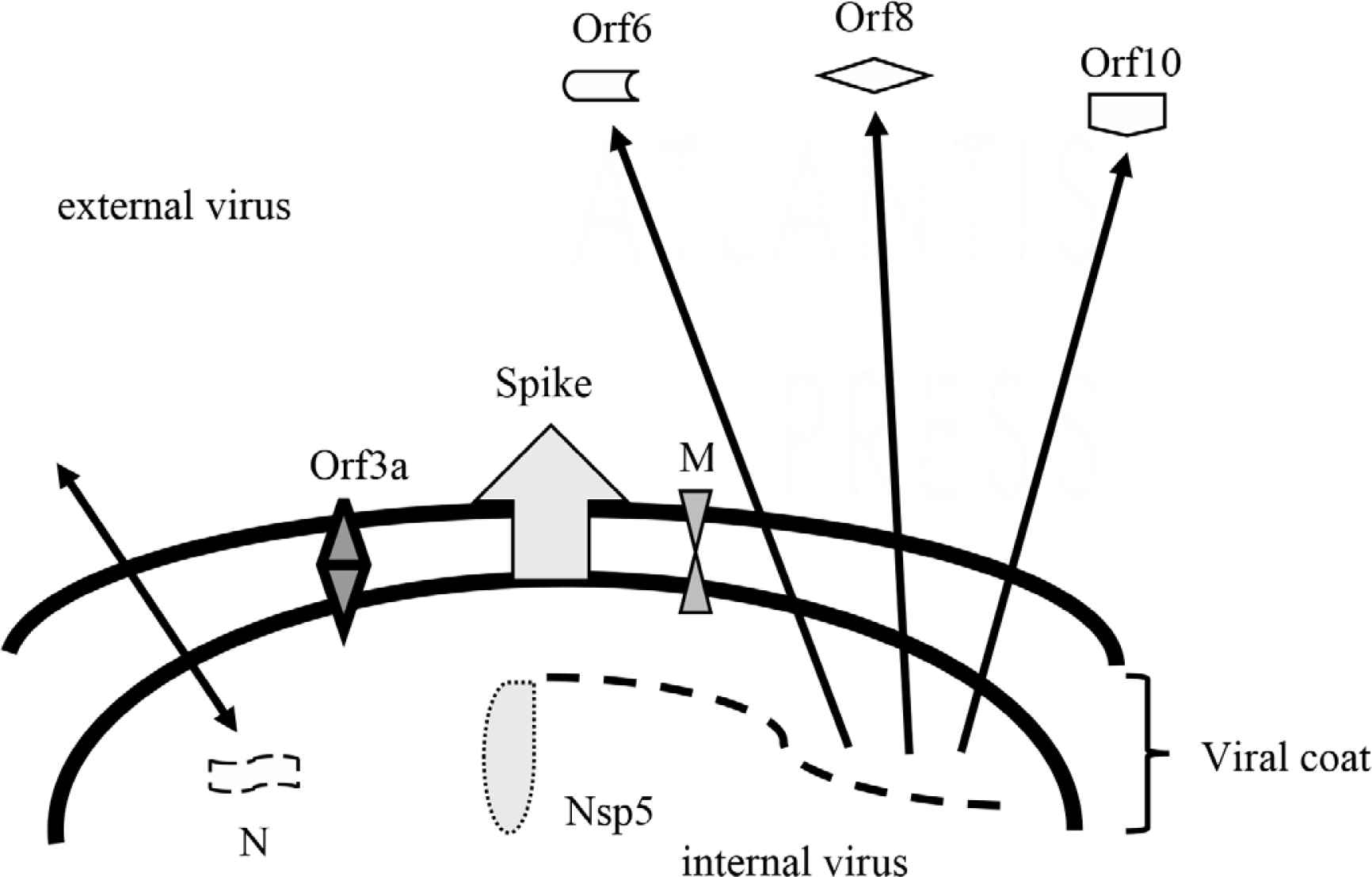
Provisional structural and non-structural immunogens of SARS-CoV-2. N, nucleocapsid protein functions in virus packaging and transcription complex; Nsp5, non-structural protein 5 also referred to as main protease [Mpro or 3CLpro] functions to process several virus proteins; M, membrane protein functions to promote intracellular virus assembly; Spike, spike protein including RBD functions in virus attachment and entry; Orf3a, open reading frame protein 3a functions as an ion channel and promoter of virus budding; Orf6, open reading frame protein 6 functions in cellular and extracellular immunomodulation; Orf 8, open reading frame protein 8 functions in cellular and extracellular immunomodulation; Orf10, open reading frame protein 10 possibly functions in cellular immunomodulation but its role is controversial if any.
Whether for enzyme immunoassay-detected antibody (or similar) or neutralizing antibody, the total specific antibody level peaks after 3–10 weeks [30,51–60]. As anticipated, IgM reactivity diminishes over the first few months while IgG reactivity continues to be detected for a much longer interval [54,56,57,59,61–67]. The finding of rapid IgG response relative to IgM suggests a possible priming from past exposure to endemic respiratory coronaviruses [34]. Circulating IgA responses generally parallel those of IgG although not as prolonged [56,62,64,65,68–70]. One study found considerable IgA titres associated with severe respiratory disease [71]. Others have found no consistent IgG/IgA correlation for intensity of production [34]. Quantitation by most methods increase with the severity of infection [33,34,36,41,50,52,56,60,70–87]. Severe disease may be associated with a delay of the humoral response [88]. Children with multisystem inflammatory syndrome amount stronger antibody responses akin to more severely ill adults, and in contrast to either children or adults with mild illness [89]. A greater quantitation of antibody at the time of disease presentation may be associated with lesser mortality [30,90]. Others have found little difference of IgG reactivity to S protein among moderate to severe infections [88,91]. Affinity maturation is time accrued [50,78]. A small proportion do not seroconvert after infection, and thus not all post-infectious sera may be useful, even though other aspects of post-infection immunity via cell-mediated immunity may have arisen in these patients [33,34,68,72–74,77,92–94]. A lack of antibody response is particularly noticeable for asymptomatic and out-patients [33,64,68,73,77,85,89,95]. Using a given threshold of anti-RBD antibody, Bartsch et al. [89] define a limit above which other immune responses and effector functions are recruited. Immunocompromise may also be a risk factor for lack of antibody production [96,97]. A portion of the latter patients may not develop IgG or neutralizing antibodies [34,35,95]. Variation in antibody production among different ethnicities, but not gender, is consistent with the genetic variation that would be anticipated [52,64,77,92]. Other authors provide evidence for a gender difference [35,58,76,80,82,86]. Patients with older age also appear to have higher antibody quantities, although they are also at risk for more severe infections [35,36,72,79,80,82,83]. Some authors have not found an age-related variance [52,64,81]. Dyssynchronous adaptive immunity may be associated with a propensity to worse disease [98,99].
Different antibody responses decay variably and dependent on the nature of the antibody target and severity of disease [33,50,54,59,61,68,75,76,78,83,87,95,96,100–103]. The latter includes measurable neutralizing antibody, and the decay in some patients after 1 month can be such that donations with desirable high titres elapse quickly in convalescence after infection [49,54,103–106]. Neutralization antibodies may decline although maturation in Fc effector functions may persist [66]. Key antibody responses are followed by memory B cell persistence [28,39,48,49,57,65,66,78,102,107]. Likewise, T cell recognition lasts well after declining humoral immunity markers [23,57,67]. The memory B potential increases with severity of infection [39]. Some groups have found a degree of anti-S2 subunit and anti-N IgG reactivity in infection-naïve individuals [45,107]. Gaebler et al. [78] have proposed that antibody maturation may be a function of prolonged antigenic exposure. Binding affinity to S is time accrued [28].
3.2. Correlations with Immunity or Protection
T cell immunity arises, as anticipated, during infection, and the quality and quantity of the same varies according to the intensity of disease. [31,48,51,63,65,67,68,70,74,89,91,93,94,98,102,109–111]. As passive immunotherapy is conventionally ascribed to immunoglobulins in a cell-free product, the translocation of such preformed cell-mediated immunity cannot be accomplished apart from immunoglobulin-related effector functions. Even if cell-mediated immunity could conceivably be ‘donated’ with alternate allogeneic blood products or stem cells, the prospect of complex incompatibility immune reactions would complicate usage. It may also be that preceding low-avidity T cell responses may commonly exist among humans, and that post-infection T cell responses may vary also in such avidity, according to disease intensity [94,109]. Mild infections may nevertheless attract memory T cell presence [23,65,102]. In a rhesus macaque model, CD8+ T cells may have a functional role even when circulating specific antibody levels are low [112]. These findings are consistent with the proposal that good quality humoral and cell-mediated responses may require a particular intensity of disease, such that the immune system must cross a particular barrier after which quality and prolongation arise [89]. Immunosuppression in animal models facilitates more virulent infection [113]. Cohen et al. [31] portray a scenario where differential T cell responses give rise to varying disease susceptibility for young and older ages. The latter is also supported by other age-related differences shown for B and T cell clonal expansions after infection [114]. These variations on age-related responses may also be superimposed on the differential character of innate immune reactivity [115].
In other systems, the contributions of both neutralizing and immunoglobulin effector functions may be relevant to the persistence of protective antiviral immunity [89,116]. That a humoral immune response occurs does not necessarily imply that such reactivity has mature effector functions [34,99]. Effector function variability in donor plasma samples is considerable [34]. Antibody effector functions can be Fc-mediated and may include such entities as antibody-dependent natural killer cell activation and degranulation, antibody-dependent cellular phagocytosis by monocytes and neutrophils, antibody-mediated complement deposition, Fc-mediated macrophage polarization, antigen presentation, and B cell activation.
There are many potential approaches to detecting neutralizing antibodies, and there are many more variations that are technically possible (Table 2) [117–119]. It is more critical to establish correlates of any such neutralization with actual protection or treatment because of potential concerns over veritable markers for the latter [118]. Whereas correlations between neutralizing antibody measures are generally good, there are nevertheless important subtle differences, and some measures may consistently show higher surrogate neutralization titres than more conventional and historic assays [119]. The presence of neutralizing antibodies as measured by pseudo-typed lentivirus particles correlated with protection [120]. It is not clear if pan-coronavirus antigen reactivity correlates with protection even though it may be associated with neutralization [45]. Lv et al. [121] proposed that SARS-CoV-1 and SARS-CoV-2 may have cross-reactive antibody to the S protein but not necessarily cross-neutralizing antibody. From studies with human monoclonal antibodies, it is apparent that in vitro measures of neutralization do not always correlate with in vivo efficacy of relative potency [122].
| Methods | Approach | Methodological nuances |
|---|---|---|
| Live virus neutralization | Live virus pre-admixed with patient serum dilutions is applied to viable cell line | Cell line susceptibility variable; different reporter systems possible for measuring virus infection; requires viable SARS-CoV-2 with inherent biosafety issues; non-cytopathic effect reporter systems possible facilitating through-put; lesser susceptible cell lines can be transduced with ACE2 or TMPRSS2 receptors to enhance assay |
| Cytopathic effect | ||
| Plaque-reduction | ||
| Focus-reduction | ||
| Pseudo-type neutralization | Alternate virus with SARS-CoV-2 antigen is pre-admixed with patient serum and applied to detector cell line; reporter signal detected variably | Non-infectious for SARS-CoV-2 thus avoiding many biosafety concerns; detector cell lines can be transformed to enhance assay; many potential forms of detector signals; high through-put possibilities |
| Vesicular stomatitis virus | ||
| Lentivirus | ||
| Murine leukemia virus | ||
| HIV-1 | ||
| Surrogate neutralization | SARS-CoV-2 attachment protein-bound surrogate is directly pre-mixed with patient serum or assessed in a competitive immunoassay; reporter signal can vary | Non-viral surrogate approach markedly lessens biosafety concerns; attachment protein can have various conformations or lengths; very amenable to high through-put; attachment nullification simplifies concept of neutralization whereas live virus method may be assessing multi-modal virus attachment and non-attachment inhibitions |
Key methods for determining SARS-CoV-2 neutralizing antibody
The presence of anti-viral IgM can correlate with neutralization potential [34]. Klingler et al. [123] suggest that IgM and IgG1 contribute considerably to neutralization and lesser so for IgA. In developing mucosal IgA, the circulating serum antibody does not necessarily have to be robust [71]. Dimeric mucosal IgA has greater potency to neutralize than monomeric IgA [69]. There is a general trend for an association of seropositivity with a lesser opportunity for apparent repeat infections over time, although these are preliminary observations [124]. Shields et al. [125] found a pre-vaccination high seroprevalence of anti-S antibody among dental professionals. A particular IgG EIA threshold was found to correlate with protection against subsequent infection and seropositivity was retained for many months.
Anti-S antibody levels correlate well with those of neutralizing antibodies, including anti-RBD [30,33,38,48,62,64,81,86,88,90,106,126–132]. Piccoli et al. [50] have proposed that some 90% of neutralizing activity is due to anti-RBD antibody, and they have particularly mapped two dominant subepitopes. Wendel et al. [105] found good correlation of anti-N antibody and neutralization. Among convalescent plasma from a voluntary donor program, a correlation was found for high IgG anti-N titres and neutralization titres ≥1/160 [105]. Another study found that both moderate and high EIA anti-N antibody correlate with neutralization titres >1:80 [43]. Others found a modest correlation of anti-S1 antibody as detected with a commercial EIA to neutralization titres >1:100 [133]. Salazar et al. [33,129] found a better correlation between anti-RBD response and neutralization titres in convalescent patient samples. Bryan et al. [134] determined that the presence of anti-N IgG correlated with a reduction in 30-day mortality. Secchi et al. [37] found a correlation of anti-RBD IgG with survival. Antibody avidity matures after infection and is correlated with duration of infection and higher neutralizing antibodies [28,135]. The avidity of anti-S correlates better than anti-N. Li et al. [36] found an association of low IgG anti-S, anti-RBD, and anti-N with a longer duration of viral RNA shedding. Antibody mapping has the potential to further define relevant antigens [136–138]. Microarrays offer a more complex mechanism to define immunoreactive epitopes even when honed down to a few larger but common antigens such as S and N proteins [42,139]. Neutralizing antibody levels may also correlate well with particular adaptive T cell responses [111].
3.3. The Potential Perils of Neutralization Escape
The D614G mutation in the S protein did not associate with neutralization escape [39,63,73]. Variant mutations of concern have recently included at least UK B.1.1.7, South Africa B.1.351, and Brazil B.1.1.28.1 [140–150]. Other groups have more recently shown that escape mutants are time accrued in tissue culture or animal models, have particular mutations, and may completely escape neutralization [150,151]. Such mutants may develop over the course of an infection [152]. Mutants can potentially escape neutralization by some but not other convalescent sera, and it is thus evident that an individual’s repertoire of antibody production should be identified; this would have bearing on the use of single donor plasma donations and their variable efficacy [153–156]. It may also be possible that the exposure to donor antibody may select further for viral mutants [157]. Likewise, resistance to therapeutic monoclonal antibodies may also arise [144,158]. In some elaborate studies of human monoclonal anti-RBD antibody, specific mutations did not affect neutralization efficacy [159]. Yet more contemporary studies suggest that mutations for S protein, including the RBD, can be constructed in vitro which do indeed have the potential to escape antibody recognition and neutralization [160,161]. A choice of more conserved targets or products with more than one target have the opportunity to overcome any such future dilemma [162,163].
The goals of an effective passive immunotherapy would preferentially include activity to key exposed and targeted viral antigens, sufficient antibody, ease of administration, persistence, resilience to escape mutation, and capability to enact effector functions.
3.4. Antibody-dependent Enhancement
For convalescent plasma, IV immunoglobulins (IVIG), or purified antibody (monoclonal or not), there remains the possibility that other beneficial or harmful effects could follow administration that are outside the role of protective immunoglobulin. The use of blood products to modulate immunity specifically in the setting of immunological storm attracts interest [164]. Likewise, there is a time-honored use of plasma for effecting compensatory physiological changes such as for severe hypoalbuminemia and fluid third-spacing dynamics.
In theory, antibody-dependent enhancement (ADE) reflects the potential of virus-specific antibody to promote the disease or create other complications [7,165]. Vaccination with various viruses in animal models best illustrates this phenomenon. Yang and colleagues raised concern about viral entry enhancement in the context of SARS-CoV-1 [166]. To some extent, similar clinical effects may have been seen in past human vaccine trials [7]. Most such effects, whether experimental or clinical, appear to occur after reinfection or vaccination, but have largely either not been seen or looked for in the context of passive immunotherapy [7]. It is also not clear that any such event, categorized in preliminary form as ADE, attracts similar clinical consequences or has the same pathogenesis [167]. From a purely in vitro perspective, there are some methods to gauge the potential for ADE to occur [159,168]. While it is yet an issue that attracts attention, the prospect that any such categorized events will prove to complicate SARS-CoV-2 immunotherapy is becoming less likely, as the experience progresses. Such concerns nevertheless may transfer to in vitro re-creation of antibodies (monoclonal or purified plasma) which have modifications of effector function domains. In this context, Zhou et al. [131] have suggested that non-overlapping RBD epitopes may attract differential immune responses which determine whether a humoral immune outcome will foster virus neutralization or ADE.
3.5. Monoclonal Antibody Derivation and Applications
If passive immunity confers a beneficial effect, the potential to harness protection through individual antibodies or pools thereof becomes an attractive proposition, especially when a non-human donor source is available at high production capability. As reviewed by Walker and Burton [169], the derivation, modification, and application of monoclonal antibodies are yet in their infancy, despite the vast research that has been conducted thus far. This also applies to their application in the field of coronavirology. Atyeo et al. [170] illustrate how modifications can affect pathological outcomes. Yet, others describe how human monoclonal antibody derivation can vary on the basis of the maturation that occurs in memory B cells [78].
One study found key neutralizing antibodies directed to RBD, N-terminal domain, and quaternary S structures otherwise [171]. Human monoclonal antibodies to RBD have been assessed singly or in tandem [12,122,159,172]. Others have examined cocktails of two monoclonal antibodies which recognize non-overlapping epitopes of the S protein [173]. Anti-S human monoclonal antibody produced neutralization in a hamster model [113].
Neutralizing murine monoclonal antibodies have been produced which target RBD [168]. These were thereafter ‘humanized’ into a chimeric antibody through the combination of the murine V region to human IgG1/kappa. The revised antibody retained neutralization capacity. In general, the production of humanized anti-SARS-CoV-2 monoclonal antibody has promise and has quickly progressed to human trials.
3.6. Animal Models of Passive Immunity
Variations of animal models used to assess passive immunity for SARS-CoV-2 infections and their implications for human treatment are detailed in Table 3. Emergent themes include an antiviral effect of convalescent serum which includes both pre-infection protection and amelioration of active disease. Effects are seen to impact on viral replication and pulmonary histopathology. The latter carries through to post-vaccination donor serum or purified IgG. Efficacy has been largely dose-dependent. These studies supported the ability of humoral immunity transfer to be of clinical value outside of the role of innate or cellular immunity, and give way to credibility for monoclonal antibodies to work singly or in combination.
| References | Model system | Treatment | Outcomes |
|---|---|---|---|
| [12] | Murine | Bioengineered human anti-RBD 200 mcg intraperitoneal | Both protection and treatment efficacy |
| [38] | Murine | Human MAb anti-N terminal S protein; 200 mcg of single or dual antibodies intraperitoneal | Protective effect |
| [112] | Macaque | Convalescent macaque donor purified IgG; 2.5–250 mg/kg for prevention and 25–250 mg/kg for treatment both intravenously | Dose-dependent for prevention, but higher dose only effective in treatment |
| [113] | Hamster | Human MAb 30 mg/kg subcutaneous | Protective for immunocompetent and immunosuppressed animals |
| [122] | Murine and hamster | Human MAb anti-RBD intraperitoneal; single 8 mg/kg or dual 1.8–16 mg/kg | Both protection and treatment efficacy; Fc functions relevant to outcomes |
| [159] | Murine and hamster | Human MAb 2–36 mg/kg intraperitoneal | Both protection and treatment efficacy |
| [168] | Murine | Two humanized murine MAb 20 mg/kg intraperitoneal four hours post-challenge | Decreased viral load in lung and associated histopathology |
| [170] | Murine and hamster | Human MAb anti-RBD modulated for Fc functions; murine 200 mcg and hamster 5 mg/kg intraperitoneal | Improved treatment outcomes for native MAb but enhancement of disease with some Fc engineered-variations |
| [172] | Murine and rhesus monkey | Human MAb anti-RBD single or double; murine 200–400 mcg intraperitoneal and monkey 50 mg/kg intravenous | Decreased viral load in lung and associated histopathology; combination of antibodies additive |
| [173] | Hamster and macaque | Human MAb REGN-COV2 combination; hamster 50 mg/kg and macaque 0.6–150 mg/kg intravenous | Both dose-dependent protection and treatment efficacy |
| [174] | Hamster | Hamster convalescent serum 2 mL intraperitoneal | Inhibited viral replication |
| [175] | Murine | Post-vaccination donor serum 600 mcl intraperitoneal; animals immunized with S-carrier virus | Reduced lung histopathology |
| [176] | Murine | Human convalescent plasma intravenously | Diminished lung histopathology and prevented mortality; dose-dependent effect |
| [177] | Murine | Human MAb 20 mg/kg intraperitoneal given a few hours after challenge | Prevented disease and benefitted active treatment |
| [178] | Murine | Human MAb anti-RBD 1 mg intraperitoneal | Reduced lung viral load and reduced mortality; dose-dependent |
| [179] | Murine and hamster | Human MAb anti-RBD 0.4–10 mg/kg intraperitoneal | Both protection and treatment efficacy; murine dose-dependent; Fc modulation affects treatment use but not protective capacity |
| [180] | Hamster | Human MAb 8 mcg–2 mg intraperitoneal | Prevented disease; prevention correlated with circulating antibody levels |
| [181] | Rhesus monkey | Single human MAb anti-RBD given intravenously; 20 mg/kg once for prevention and 50 mg/kg twice for treatment | Both protection and treatment efficacy |
| [182] | Murine | ‘Bi-specific’ engineered non-overlapping anti-RBD; antibody derived from two human MAb; 150 mcg given intraperitoneally pre-challenge | Prevented disease |
| [183] | Murine, hamster, and macaque | Human MAb anti-RBD; 10 mg/kg intraperitoneal for rodents and 10 mg/kg for macaques | Preventative and therapeutic for mice; reduced viral load in hamsters; reduced viral load and lung pathology in macaques |
MAb, monoclonal antibody; RBD, receptor binding domain of Spike protein; S, Spike.
Key contributions for animal models of passive immunity in SARS-CoV-2 infection
Considerable work has been done with monoclonal antibodies, especially human-derived. Again, there are variable benefits both for prophylaxis and active treatment in a dose-dependent manner. Reductions are seen in both viral load and lung disease. A multiplicity of monoclonal antibodies can perform better than a single one. Such a strategic tactic was able to protect while giving promise to avoid escape mutations. Most such antibodies are targeted to the N-terminal of the S protein or the RBD. Effector functions of the antibody Fc fragments can either improve or complicate infection and, thus, considerations on best contour of that region of the antibody are important. There is potential for such immunity to augment protection for immunocompromised patients.
3.7. Human Trials of Passive Immunity
Human monoclonal antibody LY-CoV555 (bamlanivimab) was trialed among outpatients with mild to moderate infections, and the investigators reported an interim analysis [184]. Patients were randomized to one of three (700, 2800 or 7000 mg) single intravenous doses. The primary endpoint of viral load was the lowest for the 2800-mg dose at day 11, but overall the differences were not dramatic. There was a trend for lower severity of illness among recipients compared to placebo, and there were trends for lower hospital need and visits to an emergency room. Further studies have used similar doses for randomized placebo-controlled trials among mild to moderate infections with LY-CoV555 and LY-CoV016 (etesevimab) [185]. A reduction in viral load was seen, but the effects for secondary outcome measures were variable.
Pre-pandemic IVIG batches from both Europe and the USA did not find significant neutralization titres to SARS-CoV-2, despite the presence of considerable neutralization activity for endemic respiratory coronavirus 229E [186]. In contrast, other IVIG sources from donations collected elsewhere yielded lots which variably neutralized SARS-CoV-1 and SARS-CoV-2, but not MERS-CoV [187]. The latter investigators also found other sources with antibody cross-reactivity to all three of the latter coronaviruses [188]. As the pandemic progresses, there has been considerable improvement in the degree of anti-SARS-CoV-2 seropositivity in IVIG lots, presumably reflecting the seroprevalence that accumulates [189]. Huang et al. [190] retrospectively reviewed cohorts with and without IVIG administration and did not find a beneficial effect. The latter study did not adjust for increased age or increased morbidities found in the treatment group and the dosing was variable (10–20 g/day for 3–5 days). The source of IVIG was not detailed (i.e., pre- or post-pandemic), and no data were provided on the presence of neutralizing or other anti-SARS-CoV-2 antibody. The benefits of IVIG are debatable, but several studies have not determined the pre-existing antiviral potency of the same [191–195]. Such studies do not preclude the potential benefit of IVIG in other niche situations, such as immune thrombocytopenia, ascending polyneuropathy, or multisystem inflammatory disorder attributable to COVID-19 [196–198]. On this theme, however, there is yet potential for the creation of IVIG specifically from donors with convalescent COVID-19 infection. Such ‘super’ infusions have the theoretical appeal to concentrate what might be beneficial from convalescent plasma otherwise. This concept also allows donor sera of lower neutralization capability to be concentrated, thus allowing for expansion of a donor pool. Concentration of convalescent sera in this fashion may require a considerable number of donor samples.
The development of convalescent donor plasma programs for COVID-19 occurred in short order [199–201]. A proportion of proven-infected donors may not have measurable antibody, and some asymptomatic infections may yet be associated with significant seropositivity [200,201]. EIA or neutralizing antibody standards were provisionally set for donor units [103]. The use of donor plasma can soon outstrip the availability in highly endemic scenarios [202]. At screening, however, there is a considerable range of antibody levels [79,80,200,203–205]. There may be some non-serological reasons why prospective donors may be rejected [201]. The collection of convalescent plasma from donor systems should exclude infectiousness, given the detection by some of viral RNA in blood. Andersson et al. [206] found viral RNA in 12.7% of donor blood product, but could not find infectious virus in a small sample of the latter. Others have not found viral RNA in the donor some weeks to months among post-infection donors [207]. ABO compatibility would be prudent [208,209]. In addition, donor samples with high neutralizing capacity would seem to be the best prospects, but not all single measures of antibody will capture seroconverters [204]. In post-infectious donors, neutralizing antibody ≥1/160 was found in approximately 64% at 28 days after clinical recovery and in approximately 41% when deemed negative for virus by RNA amplification [105]. Another study found 42% of samples had neutralization titres >1:100 [133]. After defining a potential donor however, there is a relatively small window in time for continued collection of similar-titred neutralizing samples. It can generally be inferred that high-titred neutralizing donations are better than others. Bradfute et al. [126] found that different measures of recipient antivirus antibody did not change pre- and post-infusion, but relatively low-titred initially unscreened plasma lots had been used. Others have reported no major change in pre- and post-infusion antibody levels in patients despite the use of presumed high EIA-titred donations [127]. Xia et al. [210] found a correlation between plasma donor antivirus antibody quantitation and such antibody in recipient blood. Donor plasma led to increased anti-S1 and anti-RBD antibody but not anti-N antibody levels in recipients. Libster et al. [211] found increased serum antiviral IgG 24 h after infusion, but the prevailing antibody at that time did not correlate with protection. Donor sera immunoglobulin possess non-neutralization effector functions which may be of variable intensity [34]. It is also relevant to consider that donor plasma may also have other non-immune effects of a salutary nature [166].
The use of convalescent plasma has been anecdotally documented in small patient series as referenced in representative samples [212–238]. These are very difficult to gauge for therapeutic efficacy, given the patient and product variability, concomitant variation in support and other treatment strategies, and the lack of controls [239]. Despite the latter, these studies nevertheless gave some promise that obvious adverse reactions were not common and, hence, provided at least some fodder for test of concept [79]. Such studies were cautiously required especially in the context of severe disease where immunological aberrations of the infection were forefront in concerns. Subsequently, very large reviews of immediate post-infusion safety have been tabulated, and there is a low incidence of acute morbidity directly related to the transfusion product [208,239–241]. Generally, the frequency is <4% [241]. Some have proposed that infusions might be associated with a reduction in excreted viral load [242]. The latter is contrasted with the lack of association found in one study between persistence of virus RNA detection in respiratory samples and presence of circulating anti-S antibody [84].
Studies with modest to high numbers of patients have now been published for various trials of convalescent plasma prevention and treatment (Table 4). Some generalizations can be extracted, but consistent protocols, doses, and measures among studies are lacking. Given the latter, it would be considerably precarious to assess the data through meta-analyses, although there is often the temptation to do so. In a simulation analysis from sub-Saharan Africa, it was suggested that convalescent plasma therapy was associated with a 50% mortality reduction [272]. The pointed example detailed by Kemp et al. [157], albeit in a unique patient context, illustrates the potential for SARS-CoV-2 to adapt to selective antibody pressures from donor plasma influence.
| References | Country | Product | Source | N | Dose | Frequency | Outcome | Comments | Side effects |
|---|---|---|---|---|---|---|---|---|---|
| Case series | |||||||||
| [126] | USA | No minimum threshold used | One donor | T – 12 | Unit (200 mL) | 1 unit | Reduced nasal viral RNA load correlates with increasing EIA or neutralizing antibody generally, but time-accrued | Open single-arm proof-of-concept; all plasma <1:160 neutralizing antibody; no change in antibody before and after plasma infusions | No reactions |
| [128] | Israel | No minimum threshold used | Single or two donors | T – 49 | 200 mL | Twice over 24 h | Clinical improvement associated with high titres and receipt <10 d. | Donors >14 days since last neg. RT-PCR and 2 neg. RT-PCR; donor units neutralizing antibody range 1:20-1-2560 | No reactions |
| [243] | USA | No minimum threshold used | Variable | T – 35, 322 | Variable | Mostly once | Lower mortality if plasma given <3 days or if plasma had high EIA antibody titres | Non-randomized, not blinded, observational study; variable time to infusion | Not detailed |
| [244] | Italy | Neutralizing Ab | Not detailed >1:80 | T – 46 | Unit | 1–3 units | Reduced mortality compared to national statistics | No direct comparative group; single-arm proof-of-concept design | Two possible transfusion reactions |
| [245] | USA | No minimum threshold used | Mainly one donor | T – 25 | 300 mL | 1–2 units | No comparison group | Safety and proof-of-concept study; donor age 23–67 years, asymptomatic >14 days, neg. RT-PCR; plasma anti-S or RBD titres 0-1:1350 | No reactions |
| [246] | Chile | No minimum threshold used | Not detailed | Early 28 | 200 mL | Twice over 24 h | No differences for several primary or secondary outcomes | Early - on enrollment and late - delayed or none given; donors asymptomatic >28 days, neg. RT-PCR, ABO matched | Two severe reactions |
| Late 30 | |||||||||
| [247] | USA | EIA IgG >6.5 arbitrary units | Not detailed | T – 38 | 200 mL | Once or twice 1–2 h apart (86.4% had 2) | Early treatment group had less mortality and less hospital stay | Donors neg. RT-PCR, ABO typed; early group had plasma after 4.6 days average and late group average 16.4 days | One possible transfusion reaction |
| [248] | USA | No minimum threshold used | Not detailed | T – 31 | Not detailed | Not detailed | 27% mortality | Observational proof-of-concept | Not detailed |
| [249] | Argentina | EIA anti-S/RBD IgG >1:40 | Not detailed | T – 4719 | 200–250 mL | 1–2 units | Early transfusion associated with reduced mortality | Retrospective analysis of all plasma recipients; advanced disease; donors RT-PCR neg.; no difference for mortality for recipient units < or >1:1600 titre | Not detailed |
| [250] | USA | No minimum threshold used | Not detailed | T – 44 | Not detailed | 1–2 units | Deceased recipients received plasma with lower antibody | Retrospective observational study; post-transfusion neutralizing antibody elevated in 5/8 patients observed after 3 days | Not detailed |
| Cohort studies | |||||||||
| [127] | USA | EIA anti-S IgG >1:1000 | Not detailed | T – 90 | 200 mL | 1 unit | No reduction in O2 need or mortality overall; lower O2 need and mortality for patients <65 years | Overall analysis and propensity score-matched analysis; donor asymptomatic >14 days and ABO match; low pre-transfusion antibody associated with greater mortality | No reactions |
| C – 258 | |||||||||
| T – 73 | |||||||||
| C – 73 | |||||||||
| [210] | China | No minimum threshold used | Not detailed | T – 138 | 200–1200 mL | Not detailed | Overall possible improvement for symptoms and mortality | Non-randomized, retrospective study; donors ABO matched; plasma given 18-40 days after symptom onset | Three minor reactions |
| C – 1430 | |||||||||
| [251] | Iran | Minimal EIA threshold used | Donor plasma pooling not detailed | T – 115 | 500 mL | Once, repeated in 24 h if no initial response | Reduced mortality, reduced hospital days, reduced days of intubation | Non-randomized, donors 18–60 years, asymptomatic >14 days, neg. RT-PCR | One mild fever and chills |
| C – 74 | |||||||||
| [252] | USA | No minimum threshold used but 90% had EIA IgG anti-RBD >1:1350 | Not detailed | T – 316 | ‘Unit’ | Once or twice | Lower mortality, less intensive care need, increased clinical improvement if given <72 h and EIA IgG >1:1350 | Interim analysis; propensity score-matched controls; donor age 18–65 years., asymptomatic, neg. RT-PCR | Not detailed |
| C – 251 | |||||||||
| [253] | Turkey | No minimum threshold used | Not detailed | T – 888 | ‘Up to 600 mL’ | Not detailed | Reduced intensive care duration, ventilator support, and vasopressor requirements; no case fatality difference | Retrospective case-control; very late administration did not associate with benefits | Not detailed |
| C – 888 | |||||||||
| [254] | USA | EIA IgG >1:320 | Single donor | T – 39 | Unit (250 mL) | Twice | Less oxygen need after 14 days, improved survival | Propensity score-matched 1:2 or 1:4; no correlation of antibody quantitation and outcomes | No reactions |
| [255] | USA | No minimum threshold used | Not detailed | T – 64 | Unit | 1–2 units | No reduction in mortality or length of hospital stay overall; increased hospital discharge if >65 years and received high titre multiple infusions | Non-randomized; case-control | Two possible transfusion reactions |
| C – 177 | |||||||||
| [256] | USA | Highest EIA titre | Not detailed | T – 341 | Unit (300 mL) | One or more (79% one) | Lower mortality if plasma given <72 h and if EIA titre >1:1350 | Propensity score-matched controls; donor ABO compatible (see reference [179]) | 2% transfusion reactions |
| C – 594 | |||||||||
| [257] | USA | No minimum threshold used | Not detailed | T – 20 | ‘Unit’ | Once | Reduced mortality rate | Non-randomized; donors age 29–79 years, >28 days asymptomatic; variable EIA-IgG donor plasma titres | No reactions |
| C – 20 | |||||||||
| [258] | Qatar | No minimum threshold used | Not detailed | T – 40 | 400 mL | Once | No difference for respiratory support requirement, viral clearance, or mortality | Retrospective case control; no antibody determinations performed on plasma units; donor asymptomatic >14 days, neg. RT-PCR, ABO matched | Not detailed |
| C – 40 | |||||||||
| [259] | Kuwait | Qualitative IgG EIA positive | Not detailed | T – 135 | 200 mL | Twice (79.3%) | Clinical improvement and lower mortality | Non-randomized, prospective, disease stratification matched; donors – clinical recovery, IgG EIA positive, ABO matched | No serious reactions |
| C – 233 | 200–400 mL | Once | |||||||
| [260] | China | EIA IgG anti-S and anti-N >1:160 | Single | T – 39 | 100–200 mL | Once | Improved clinical outcomes for three comparisons (all patients diabetic) | Retrospective case control; donor 18–55 years, >1:160 EIA IgG, ABO matched; performed two propensity-matched analyses for moderate/severe and mild infection groups | No reactions |
| C – 328 | |||||||||
| T – 39 | |||||||||
| C – 39 | |||||||||
| T – 29 | |||||||||
| C – 29 | |||||||||
| [261] | Poland | No minimum threshold used | Not detailed | T – 55 | 200–267 mL | Once or twice | Shorter hospital stay with early treatment <7 days; no difference for most end-points | Non-randomized, retrospective; three analyses with different control and treatment groups | No reactions |
| T – 78 | |||||||||
| C – 236 | |||||||||
| C – 715 | |||||||||
| [262] | USA | EIA total IgG >1.4 commercial assay | Single or two donors | T – 29 | 220 mL | Twice | Less ICU transfer and reduced mortality at 28 days; not statistically significant | Non-randomized; matched controls; patients had severe infection | No serious reactions |
| C – 48 | |||||||||
| [263] | USA | Not determined | Single or two donors | T – 35 | 200–250 mL | Once or twice | No difference for length of hospital stay or mortality | Non-randomized, propensity score matched 1:2; severe infection; donors ABO matched, asymptomatic × 14 days, and RT-PCR negative | No transfusion reactions |
| C – 61 | |||||||||
| [264] | Philippines | Not determined | Not detailed | T – 75 | Not detailed | Once | No difference for length of hospital stay or mortality | Non-randomized; moderate to severe disease; controls age, gender, and diseased matched | One mild transfusion reaction |
| C – not detailed | |||||||||
| [265] | USA | Not determined | Not detailed | T – 47 | 200 mL | 1–3 transfusions | No difference for 7 days composite or mortality | Non-randomized; matched controls taken from pre-plasma use era; not all recipient patients used (47 of 94); donors ABO compatible; plasma given mean 4.9 days post-admission; recipients had severe disease | Not detailed |
| C – 47 | |||||||||
| [266] | Saudi Arabia | Not determined | Not detailed | T – 40 | 200–400 mL | Discretionary numbers up to 5 | Lower mortality; no difference for days ventilated or to clinical recovery | Non-randomized; propensity score matched 1:3; donors ABO matched, >18 years, positive for ‘rapid serology’, clinical recovery and RT-PCR neg. | No transfusion reactions |
| C – 124 | |||||||||
| Prospective randomized studies | |||||||||
| [211] | Argentina | EIA anti-S IgG >1:1000 | Single | T – 80 | 250 mL | Once | Reduced progression to severe disease and longer time to severe disease | Double-blind, placebo-control; donors - asymptomatic for 3 days, neg. RT-PCR; plasma given <72 h after onset of illness; dose-dependency; trial stopped | No reactions |
| C – 80 (mean age ~77 years) | |||||||||
| [267] | China | Minimal EIA threshold used | Donor plasma pooling not detailed | T – 52 | 4–13 mL/kg; median 200 mL | Once | No significant difference for 28 days clinical improvement, 28 days mortality, or time to hospital discharge | Randomized, not blinded; donors 18–55 years, asymptomatic >14 days, neg. RT-PCR × 2; plasma given 30 days median interval between onset and randomization; trial stopped | Two possible transfusion reactions |
| C – 51 | |||||||||
| [268] | India | No minimum antibody threshold | 1 or 2 donors | T – 235 | 200 mL | Twice over 24 h | No significant difference for severe disease progression or 28 days mortality | Randomized, not blinded; donors 18–65 years, asymptomatic for 28 days, negative RT-PCR × 2; median donor neutralizing antibody 1:40 | 1% transfusion-related side effects |
| C – 229 | |||||||||
| [269] | Spain | Minimal EIA threshold used | 1 donor | T – 38 | 250–300 mL | Once | Decreased progress to mechanical ventilation or death; no significant difference for days for oxygen or in hospital or days of mechanical ventilation or change in clinical ordinal scale; no difference for blood viral RNA (small no.) | Randomized, not blinded; EU requirements for donation, asymptomatic >14 days, all donor plasma neutralizing antibody >1:80, median 1:292; trial stopped | Two possible transfusion reactions |
| C – 43 | |||||||||
| [270] | Iraq | EIA IgG >1.25 ratio | Not detailed | T – 21 | Not detailed | Not detailed | Less duration of infection and less mortality | Randomized; donors <50 years; plasma given mean 14 days after onset; better outcome with high titred plasma | One possible transfusion reaction |
| C – 28 | |||||||||
| [271] | Netherlands | Neutralizing antibody >1:80 | Not detailed | T – 43 | 300 mL | Once or twice (second could be given after 5 days) | No difference for mortality, hospital stay, or day 15 disease | Randomized; donors ABO matched, asymptomatic >14 days; trial stopped | No reactions |
| C – 43 | |||||||||
| [4] | Argentina | EIA total IgG >1:400 | 1 donor or pool of 2–5 donors | T – 228 | 400–600 mL | Once | No difference for clinical outcome ordinal or mortality | Double-blind, placebo-control; donors 18–60 years, neg. RT-PCR, 28 days asymptomatic; median donor plasma EIA total IgG 1:3200/neutralizing antibody 1:300 | 5% infusion-related events |
| C – 105 | |||||||||
N, patient number; T, treatment; C, control; RT-PCR, reverse transcriptase polymerase chain reaction diagnostic; EU, European Union; EIA, enzyme immunoassay.
Human studies of convalescent plasma treatment for COVID-19
The concept of therapeutic plasma exchange followed by convalescent plasma therapy has been discussed [273]. However, it is unclear how removal of potentially pre-existing antibody may juxtapose with subsequent donor immunity and final outcomes. Another unique but related approach has been piloted by Selzman et al. [274] with the use of therapeutic donor human amniotic fluid. Given intravenously, there is the potential for immunomodulating effects on active COVID-19, but when donated by a convalescent mother who suffered active infection prior to birth, there is also the potential for transfer of maternal virus-directed IgG for a form of passive therapy.
Overall, the findings thus far suggest a dose-dependency and a correlation with better outcome for higher doses of administered donor plasma. The latter approach must also weigh the complicating factors of volume expansion in the short term, if higher doses are met with the need for larger transfusions. A time-dependency with regards to stage of illness also emerges. Repeat donations will need further evaluation. The definition of protective correlates continues to be a high priority.
4. CURRENT PROSPECTS
In 2020, emergency use authorizations for convalescent plasma in COVID-19 treatment were being adopted throughout the world. The latter was occurring in a context of relatively limited knowledge but also within a void of effective therapeutic prevention and treatment strategies. In the passion to create immediate and effective products, several aspects of passive immunotherapy have taken somewhat lesser priority.
The pharmacokinetics and immunokinetics of any passive antibody administration are considerably variable among patients. Whether given intravenously or subcutaneously, the volume of distribution is relatively small as a first central compartment. While the antibody may remain in circulation for a variable period of time, entrance into tissue, especially interstitial spaces, follows soon after. Much of this secondary compartmentalization will include sites where viral replication does not necessarily occur. Many of these sites will attract more antibody only due to the inflammatory nature of plasma egress. Given the large molecules of immunoglobulins, degradation immediately through liver or urinary loss are unlikely. Rather, degradation is more likely to occur with proteolytic pathways in sites of active inflammation or through usual protein handling routes. If given by subcutaneous routes, antibody is primed to migrate through lymphatic systems first, rather than direct blood circulatory paths. The handling of such protective antibody through intraperitoneal routes in animal models does not guarantee comparability for human intravenous administration. For monoclonal antibodies, the distribution will be product-specific but susceptible to several patient factors [275]. Apart from non-specific losses, antibody linking to targeted antigens and antigen-processing cells would be desired, but a localized or generalized inflammatory reaction may have various other mechanisms to attract antibody. In addition, some immediate loss may be due to the inadvertent immunogenicity of the product itself, perhaps more significant in multiple infusions. In advanced illnesses, antibody clearance may be enhanced in inverse relationship to the serum albumin concentration which may be low due to the existing biological adversity [276]. The pharmacokinetics of intravenous immunoglobulin are mainly understood from distribution to healthy patients [277,278]. It is not known how the acute inflammation during COVID-19 may alter the latter. When plasma is administered, there will be temporary expansion of the intravascular volume from redirection of body fluid, but the distribution of colloid including antibody is expected to depend on the physiological state as well [279]. For example, clinical sepsis alone can abundantly facilitate capillary leak [280]. The focus of passive immunotherapy has largely been on potency, but the factors of distribution and bioavailability for immunity drivers may be just as, if not more, important.
There is some merit to reconsidering what should have greater priority given the immediate needs of the populace. Monoclonal antibodies may be in favour for many reasons but, practically speaking, a polyclonal multiantigen immunotherapy approach makes most sense, given the potential ability to focus on multiple targets while the virus may slowly change. Multiantigen immunotherapy may also be seen as one that may target multiple exposed and newly formed viral immunogens during the breadth of the infectious process. The concept of IVIG with concentrated polyclonal antivirus activity would allow for a broader collection of donor samples even when they are of lesser whole sample quality. The latter is only a few steps away from the capability of large scale productions. Unfortunately, the latter also introduces the precarious potential for complications of pooled human transfusion. For simple donor plasma infusions, a repeat especially with different donors allows for diversity in the antiviral properties but again creates the potential for enhancing donor-related problems. If virus escape continues to be an issue, as anticipated from the analogy with the yearly behavior of endemic respiratory coronaviruses, the polyclonal approach with donor samples or monoclonal cocktails will take precedence. A polyclonal approach also has the potential to better address the multifunctional Fc effector attributes that may also contribute to the overall beneficial outcomes.
The cumulative laboratory studies suggest that prevention is possible as is active treatment. Passive immunotherapy has focused on the systemic domain but less on mucosal protection. Yet, it is abundantly clear that, in the least, earlier prevention benefits from mucosal antibody [6,7]. Maternal antibody through lactation has one of the greatest potentials for passive protection. Translating other human passive immunity into the sphere of mucosal protection is likely to be highly relevant in the understanding of the larger picture.
Proof-of-concept and open-label studies are necessary parts of progressive scientific endeavor. There is emphasis nonetheless for high quality studies which are placebo-controlled, blinded, and randomized [199,281]. Such studies are relatively complex and require considerable time and effort. While most desirable, they do not preclude lesser attempts which provide cumulative experience. The complexity of passive immunotherapy for the prevention and treatment of COVID-19 is much more than meets the eye. The need for practical and timely interventions necessarily tends to shortcut this complexity, but any failure in outcomes must not dissuade the medical and scientific community from continuing to pursue the next level of design and experimentation, because there will always be a better way. For example, the conjoined effects of immunotherapy and antiviral agents have considerable potential [282]. Passive immunotherapy should and will succeed for some patients. Rational approaches will emerge, as long as the clinical need for these products continues.
CONFLICTS OF INTEREST
The author declares no conflicts of interest.
FUNDING
Funding was not sought for this publication. There is no third party support including that from the pharmaceutical industry.
REFERENCES
Cite this article
TY - JOUR AU - Nevio Cimolai PY - 2021 DA - 2021/04/16 TI - Passive Immunity Should and Will Work for COVID-19 for Some Patients JO - Clinical Hematology International SP - 47 EP - 68 VL - 3 IS - 2 SN - 2590-0048 UR - https://doi.org/10.2991/chi.k.210328.001 DO - 10.2991/chi.k.210328.001 ID - Cimolai2021 ER -