
Dietary Flavonoids in the Management of Huntington’s Disease: Mechanism and Clinical Perspective
Additional information: Graduate Institute of Education, Department of Molecular Medicine, SANKO University, Gaziantep, Turkey
- DOI
- 10.2991/efood.k.200203.001How to use a DOI?
- Keywords
- Anti-inflammatory; antioxidant; clinical perspective; flavonoids; Huntington’s disease; intracellular pathways
- Abstract
Huntington’s disease (HD) is a neurodegenerative disorder characterized by progressive loss of neurons, which leads to behavioral systems and mental decline. HD is linked to repeat expansions of cytosine, adenine, and guanine in the Huntingtin (HTT) gene that give rise to mutation, leading to the formation of the HTT protein product. Oxidative stress also provokes the initiation and progression of HD as it leads to protein misfolding that results in the formation of inclusion which clumps together and alters neurotransmission. Despite the advancement in the field of pharmaceutical sciences, current therapeutic approaches suppress only the severity of symptoms and no therapy exists that can cure HD from its root cause. Flavonoids are the most abundant polyphenols widely present in daily dietary sources. Dietary flavonoids have a wide range of pharmacological bioactivities and many therapeutic applications. Dietary flavonoids including hesperidin, naringin, quercetin, rutin, fisetin, myricetin, luteolin, and epigallocatechin 3-O-gallate can prevent and manage HD through exerting antioxidant and anti-inflammatory activities, altering intracellular pathways, genetic alterations, and metal ion chelation. This review highlights flavonoids as therapeutic options for HD and will open new dimensions for flavonoids as safe and effective therapeutic agents in diminishing HD.
- Copyright
- © 2020 International Association of Dietetic Nutrition and Safety. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
Polyphenols, including tannins, flavonoids, stilbenes, lignans, and phenolic acids, are the most abundantly occurring secondary metabolites found throughout the plant kingdom (Figure 1) [1]. Flavonoids are widespread polyphenols in nature as more than 11,000 flavonoids have been reported so far and their daily intake varies between 20 and 500 mg, obtained mainly from fruits, vegetables, red wine, tea, and cocoa [2,3]. Flavonoids can be classified into several classes including flavonols, flavones, isoflavones, flavanones, flavan-3-ols, and anthocyanidins, depending on the oxidation level and pattern of substitution of their phenyl-benzopyran structure (Figure 1) [4].
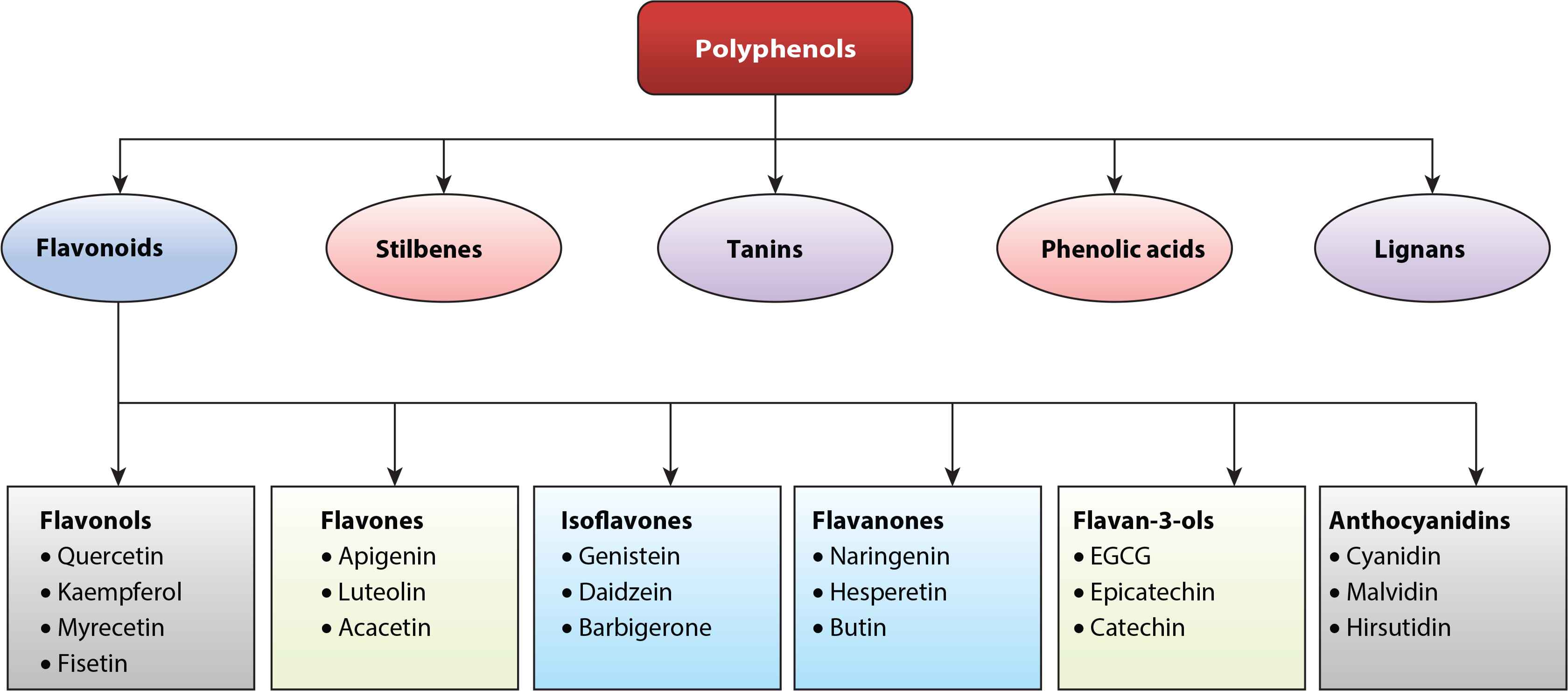
Classification of polyphenols and flavonoids with selected examples.
The chemical structure of flavonoids consists of a 15-carbon skeleton containing three rings, namely, A, B, and C (Figure 2). Rings A and B are known as phenyl rings, whereas ring C is the heterocyclic 4H pyran ring that links rings A and B together. Hydroxyl groups often occur at the C-2, C-3, C-5, C-7, C-3′, C-4′, and C-5′ positions of flavonoids. O-glycosylation with glucose, rhamnose, galactose, and arabinose often occurs at the C-3 and C-7 positions of flavonoids; however, flavonoid C-glycosides are found mainly at the C-6 and C-8 positions [5,6]. Variations in the chemical components of flavonoids affect their bioavailability and bioactivities in vivo [7].
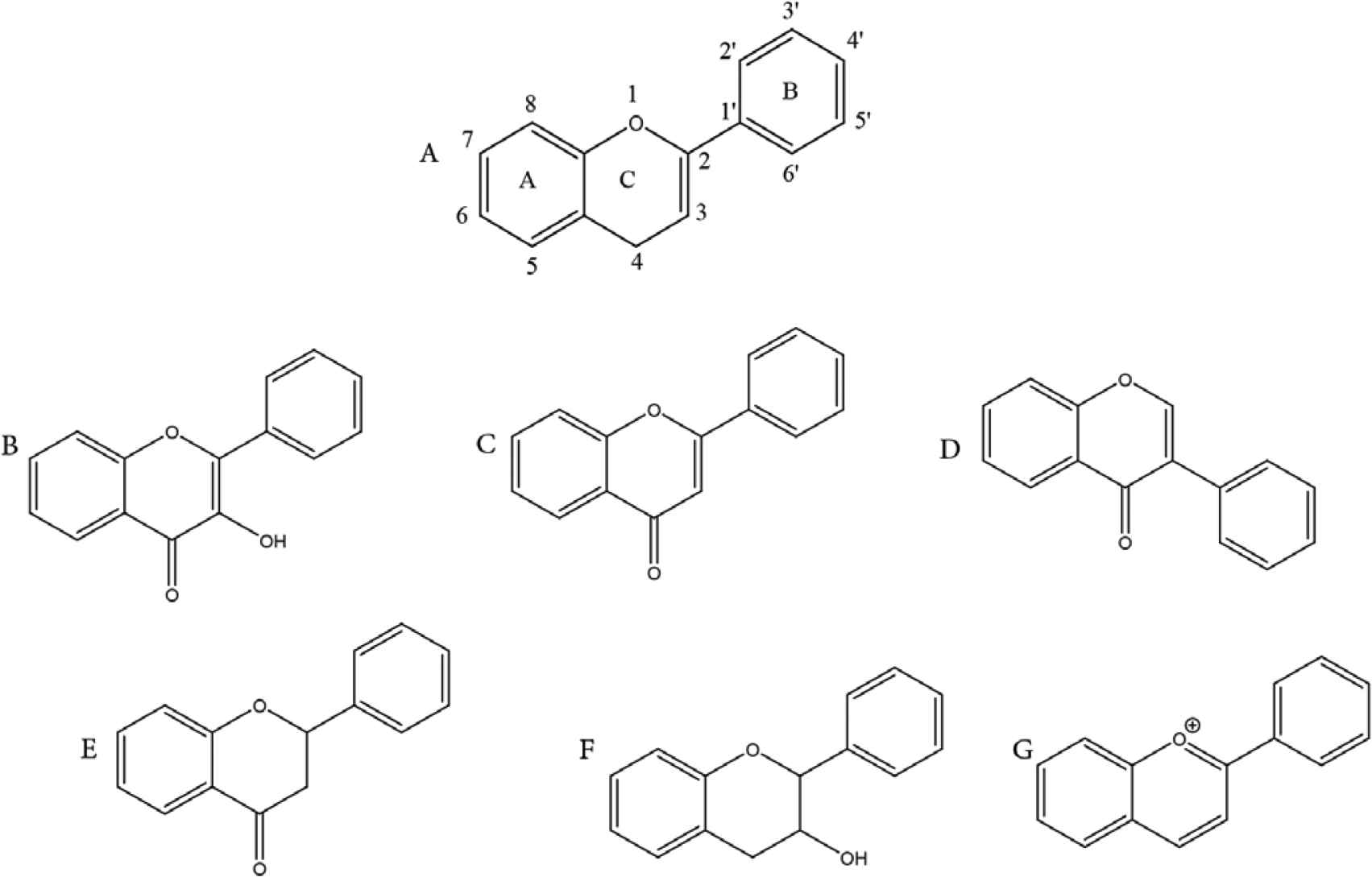
Chemical structures of flavonoids and subclasses: (A) general structure of flavonoid; (B) flavonols; (C) flavones; (D) isoflavones; (E) flavanones; (F) flavan-3-ols; and (G) anthocyanidins.
Huntington’s Disease (HD) is a progressive neurodegenerative disorder and occurs mostly from 35 years of age. HD affects muscle coordination and leads to behavioral systems and mental decline [8]. HD is linked to repeated expansions of Cytosine, Adenine, and guanine (CAG) in the Huntingtin (HTT) gene, which give rise to mutation leading to elongation of the polyglutamine tract and formation of the HTT protein product, which may aggregate throughout the brain [9,10]. Oxidative stress may also provoke HD due to the formation of Reactive Oxygen Species (ROS), which can lead to protein misfolding that causes the formation of inclusion bodies that clump together and stop neurotransmission [11]. Despite advances in the field of medical and pharmaceutical sciences, current therapeutic approaches only suppress the severity of symptoms and do not cure HD completely [12].
The role of flavonoids as neuroprotective agents has been well studied and documented in several neurological disorders such as brain edema, brain ischemia, Amyotrophic Lateral Sclerosis (ALS), brain tumors, Parkinson’s Disease (PD), and cognitive impairments [13,14]. Antioxidant potential, anti-inflammatory activity, and modulation of signaling pathways are responsible for the neuroprotective effects of flavonoids [14]. The current review highlights the therapeutic and mechanistic possibility of flavonoids in preventing and managing HD.
2. PATHOPHYSIOLOGY OF HD
Recently, many studies have shown that immune and mitochondrial dysfunction, systemic toxicity, and protein degradation are important mechanisms for HD pathophysiology (Figure 3) [15–18]. The first symptoms are usually noted between 35 and 45 years of age and death typically appears within 15–20 years after the initial diagnosis. HD is characterized by motor symptoms, psychiatric/behavioral problems, and cognitive deficits [19].
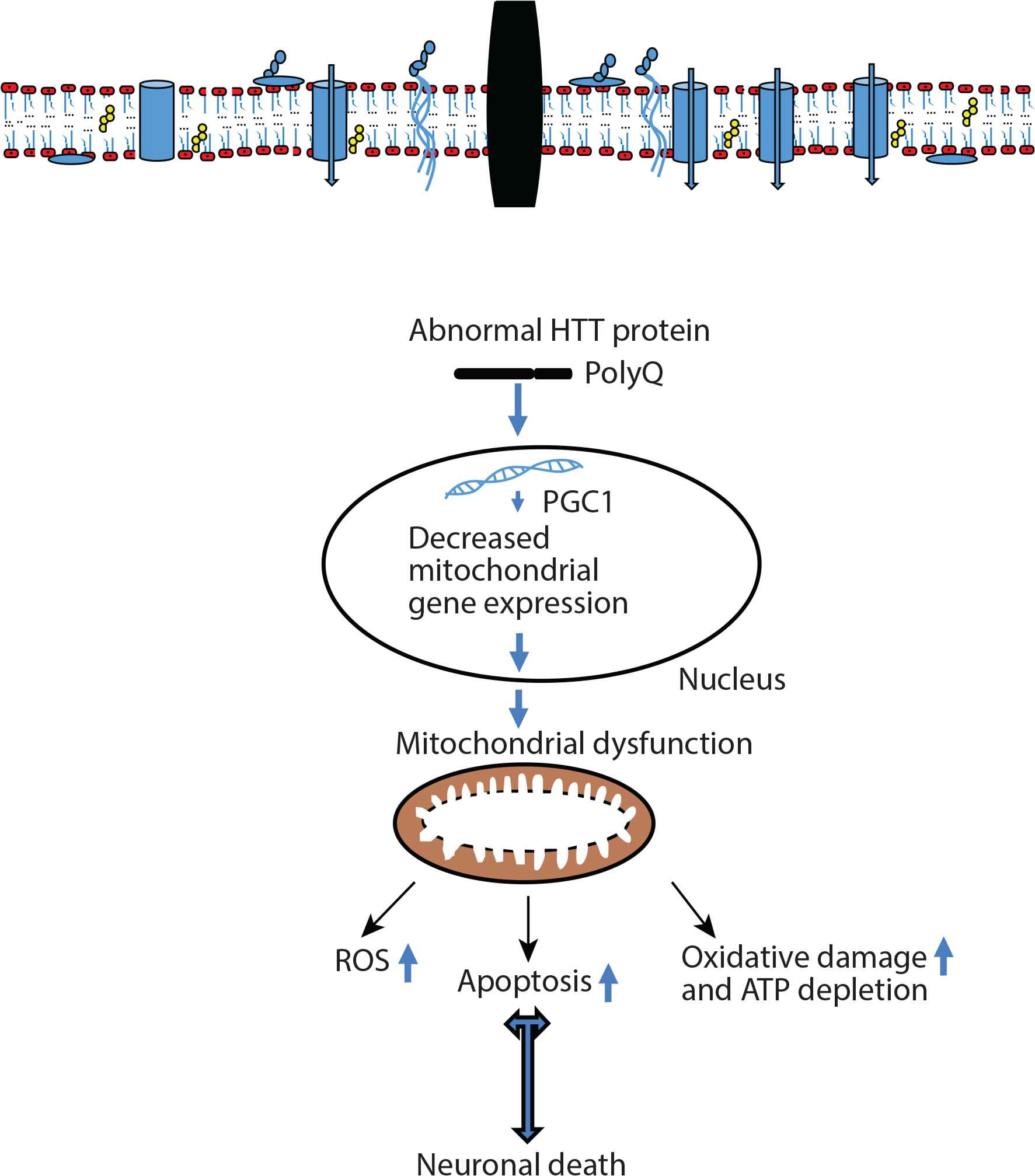
Pathophysiology of Huntington’s Disease (HD). Peroxisome Proliferator-activated Receptor-γ Coactivator 1α (PGC1α) plays an important role in mitochondrial biogenesis and mitochondrial function in neuronal cells. Abnormal HTT protein may be represented by the interference with nuclear gene transcription, thus PGC1α significantly improves as transcriptional coactivator involved in adaptive thermogenesis of mitochondrial biogenesis. HD-specific systemic toxicity mechanism mediates mitochondrial dysfunction. Mutant HTT protein response and mitochondrial stress decrease mitochondrial dysfunction through mitochondrial gene expression in the nucleus which can cause mitochondrial dysfunction. ROS, reactive oxygen species.
2.1. Huntington Protein CAG Repeats
Our current knowledge on the relevant pathophysiologic mechanisms underlying HD causal mutation remains poorly understood. The presence of the mutant HTT protein is known to be responsible for a variety of normal processes that eventually result in the death of neurons. The HTT protein is a large protein molecule consisting of many helical repeat units [20]. HD is an autosomal dominant and a highly penetrant disorder. The HTT gene is located at the 4p16.3 region of chromosomes. There are polymorphic cysteine, adenine, and guanine triplet repeats (which encode the amino acid glutamine) in the first exon of the HTT gene [21]. Repeats 1–26 are considered normal alleles and not associated with HD. Repeats 27 and 35 are called “intermediate alleles” that are not initially associated with the symptomatic disease [22]. However, during gametogenesis these repeats tend to increase and cause symptomatic disease in the next generation. Repeats 36–39 are considered “low penetrant alleles,” which carry a certain risk of HD (70% possibility) at 75 years of age; repeats 40 and over are considered “full penetrant alleles” that are responsible for causing symptomatic HD, and result from an increase in repeat numbers of intermediate alleles during spermatogenesis [22,23].
The same abnormal polyglutamine triplet repeat is also responsible for spinal and bulbar muscular atrophy, dentatorubral–pallidoluysian atrophy, and a collection of spinocerebellar ataxias (SCA-1, -2, -3, -6, -7, and -17). These neurological disorders are associated with the genomic expansion of a CAG triplet repeat in the disease-specific genes [24,25]. The resulting proteins exhibit an abnormal extension of glutamine residues to the polypeptide chain that can range from 40 to over 300 pathogenic repeats [26]. The HTT protein is known to play postdevelopmental roles in axonal and dendritic transport, regulation of gene transcription, and cell survival. At the protein level, a mutation responsible for the production of abnormally long tracts of polyglutamine (polyQ) repeats near the N terminus of HTT occurs. The HTT protein is not only primarily cytoplasmic, but also localizes to the nucleus and organelles [20,21,27]. The HTT protein expression is higher in the nervous system than in other tissues, and it has many cellular roles such as membrane targeting [28], post-translational modification [29], carbohydrate metabolism [30], iron metabolism [31], mitochondrial and endoplasmic reticulum stress, apoptosis, binding to chaperones [32], as well as providing a site for potential regulatory post-translational modifications, and the structural basis of oligomer formation. The role of these cellular functions has been mostly studied in the context of mutant polyglutamine protein [32].
2.2. Protein Degradation and Ubiquitin—SUMOylation—Nuclear Factor Kappa B
Normally, after protein synthesis, misfolded proteins that are produced as a result of different cell stress conditions are held in the lumen of the endoplasmic reticulum by chaperone proteins, thus preventing their entry into the Golgi apparatus. When these abnormal proteins accumulate in the lumen of the endoplasmic reticulum, unfolded protein response (UPR) occurs [33]. During UPR cells increase the expression of the chaperone proteins. The improperly folded proteins are discharged from the endoplasmic reticulum to the cytoplasm to be degraded in the proteasomes. Besides, persistent endoplasmic reticulum stress and an activated UPR can cause apoptotic cell death. Unfortunately, large numbers of misfolded proteins such as abnormal polyglutamine-containing HTT, which are produced as a result of increased numbers of CAG triplet repeat containing the HTT gene, are synthesized that are beyond repair. These not only cause cytotoxicity but also lead to abnormal cellular status, particularly neurodegeneration. The regulatory mechanisms for intracellular mutant HTT protein degradation may be rearranged between the ubiquitin–proteasome system and the autophagy–lysosome system [32]. Protein degradation subunits have also been detected within polyQ aggregates and may represent a cellular attempt to recruit defense mechanisms against protein misfolding and aggregation or possibly against the failure of the protein degradation system. The pathophysiological mechanisms involved in neurodegeneration processes such as protein aggregation, mitochondrial dysfunction, oxidative stress, RNA transcription are generally affected by small ubiquitin-like modifier (SUMO). The SUMO proteins default on their solubility function, which has been shown to be effective in regulating protein aggregation and in the pathogenesis of neurodegenerative diseases. However, the direct effects of SUMO protein’s solubility on α-synuclein, DJ-1, HTT, and Signal Transducer and Activator of Transcription-1 (STAT1) have also been shown [29,32,34]. SUMO proteins have been identified in inclusions in various neurodegenerative diseases such as multiple system atrophy, HD, and other related polyglutamine disorders [35,36]. The number of polyQ repeats ranges from 35 to 300, resulting in a variety of neurological diseases. Although these disorders have very different clinical and neuropathological properties, abnormal accumulation of polyQ-containing proteins and the formation of cytoplasmic and/or intranuclear inclusions in neurons have necessitated their co-occurrence as common features. Many SUMO targets have been proposed in neurodegeneration resulting from polyQ accumulation. By contrast, the presence of SUMO in inclusions in various polyQ disorders still needs to be studied in depth. Ras Homolog Enriched in Striatum (Rhes) is a small guanine nucleotide-binding protein (G protein) that is very selectively localized to the striatum. mutant HTT (mHTT) is SUMOylated, which increases the soluble form of mHTT and elicits cytotoxicity and neurotoxicity in a Drosophila model of HD [26,35].
Recently, a great number of studies have shown that extended polyQ CAG repeat triplet is important in SUMOylation [18,22,24,25]. Extended high doses of SUMO proteins because of polyQ repeats are known to suppress IK kappa B (IKκB). Thus, the inhibitory effect of IKκB is eliminated and Nuclear Factor Kappa B (NF-κB) remains constantly active. NF-κB can thus directly bind with the HTT gene promoter and increase its transcriptional activity, which causes a neurotoxic effect that could result in a detrimental increase in the expression of mHTT in striatal neurons. Given the selective physiological effects of HD pathogenesis in the striatum, there is increasing evidence indicating that the lysosome-mediated degradation pathway Macroautophagy (MA) plays a critical role in aggregate clearance [18,25]. MA is one of the important pathways of autophagy that traffics cytosolic cargoes for lysosomal degradation. NF-κB activation can also prevent MA/autophagy activation induced by inflammatory cytokines. In MA, cargoes get into a transient organelle known as autophagosome, a multilamellar structure formed through an arrangement of several proteins, including those known as the autophagy-related proteins [32].
2.3. Systemic Toxicity and Mitochondrial Dysfunction
Oxidative stress is an important contributor to several neurodegenerative diseases such as HD, which suggests that there may exist shared pathological mechanisms activated by independent and apical primary causes. HD-specific systemic toxicity mechanism mediates mitochondrial dysfunction. Mutant HTT protein response and mitochondrial stress decreased mitochondrial gene expression in the nucleus which can cause mitochondrial dysfunction. It is well-known that an important relationship exists between mitochondrial function and excitotoxicity [29,30]. More recent biochemical data have emphasized that brain tissues from HD patients showed impaired activity of respiratory chain complexes and tricarboxylic acid cycle enzymes. Thus, this impairment in normal functions because of mitochondrial dysfunction would result in decreased mitochondrial biogenesis, oxidative stress, ATP deficit, and increased apoptosis in HD disease. It is well-known that aconitase activity is particularly susceptible to regulation by ROS, reactive nitrogen species, OH, and other toxic molecules [37]. Indeed, accumulation of several markers of oxidative stress, such as lipofuscin derived from peroxidation of unsaturated fatty acids [38], increased iron metabolism [39], protein oxidation and nitrosylation [40], and 8-hydroxy-2-deoxyguanosine in mitochondrial DNA [41], is a well-described feature of HD brains. There is some evidence supporting a role for peroxisome Proliferator-activated Receptor-γ Coactivator 1α (PGC1α), which plays an important role in mitochondrial biogenesis and mitochondrial function in neuronal cells [42].
The Mechanistic Target of Rapamycin (mTOR) is a serine–threonine kinase that integrates signals to arrange cell growth and metabolism. mTOR consists of two distinct complexes: mTORC1 and mTORC2. Under nutrient-rich conditions, mTORC1 is activated and promotes protein translation and cell growth, whereas nutrient withdrawal inactivates mTORC1 and initiates MA as a cell survival mechanism. mTORC1 positively controls mitochondrial biogenesis [43] and regulates lipid homeostasis by controlling cholesterol synthesis [32,39,40]. In the striatum, Rhes serves as the main activator of mTORC1. Genetic knockout of Rhes can diminish mTORC1 activity [43]. In addition, Rhes facilitates SUMOylation [36].
3. CLINICAL STUDY FOR HD
Currently, there is no specific treatment for HD and most available therapeutics and treatment provide only symptom relief. Therapeutic options for the management of HD are summarized in Figure 4.
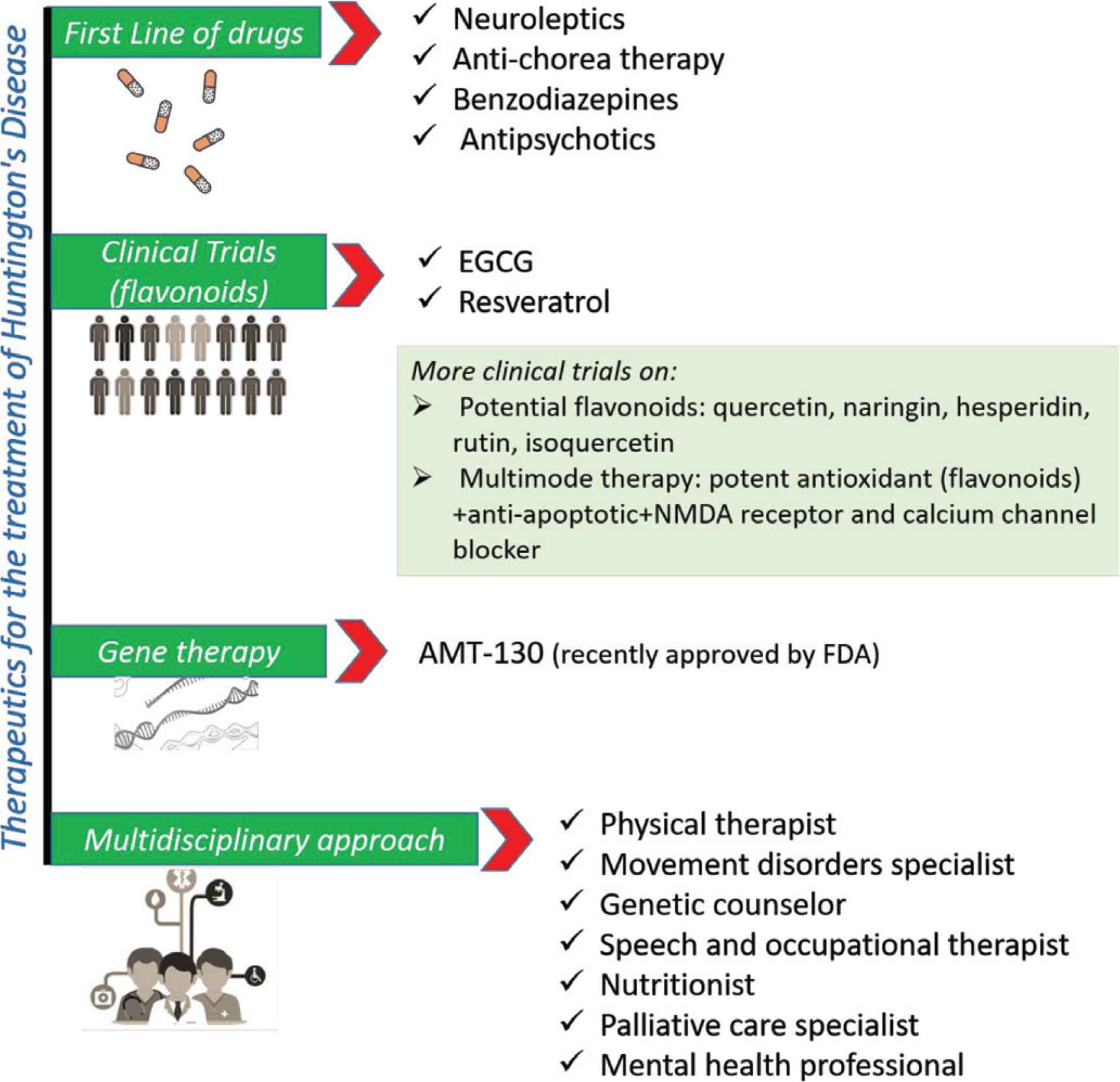
Therapeutics for the treatment and management of Huntington’s disease. FDA, Food and Drug Administration; NMDA, N-methyl-
Xenazine (tetrabenazine) and AUSTEDO (deutetrabenazine), which belong to the antichoreic class of medications, are the only Food and Drug Administration-approved drugs for HD [44]. However, their application is limited as they tend to propagate depression and suicidal attempts in patients. Various approaches have been applied for the management of HD and its associated symptoms. Neuroleptics including antidepressant, mood stabilizers, and psychostimulants are mainly used as first-line drugs for HD symptoms. In addition, multipurpose medications such as haloperidol were also prescribed to treat chorea, agitation, and anorexia. These medications provide symptomatic relief in some cases, but can also lead to several side effects and adverse effects, which lower their acceptability. Recently, a randomized, phase-controlled clinical trial of AMT-130 (gene therapy) (https://clinicaltrials.gov/ct2/show/NCT04120493) in patients with early manifest HD was designed to establish safety and proof-of-concept.
Phytochemicals, especially natural antioxidants such as polyphenols, are found to be strong candidates for preventing oxidative stress in various neurological studies [45–47]. More specifically, in neurological diseases such as HD, cell and animal model studies demonstrated beneficial effects of flavonoids in scavenging free radicals and preventing oxidative stress [48–50]. Flavonoids exerted various bioactivities against neurological diseases and reduced their symptoms in preclinical and clinical studies [51,52]. However, very limited clinical trials have been performed with regard to flavonoids and antioxidants against HD, which showed mixed results.
For example, in one clinical trial, Epigallocatechin Gallate (EGCG) improved cognition in HD patients (NCT01357681). A randomized, double-blind, stratified, placebo-controlled multicenter trial of EGCG involving 54 HD patients was conducted [53]. HD patients were administered a maximal daily dose of 1200 mg EGCG for 12 months and results were compared with placebo. Results were measured using the Unified Huntington’s Disease Rating Scale (UHDRS)-Cognition Composite Score of Stroop Test, Verbal Fluency Test, and Symbol Digit Modalities test. The results showed that EGCG improved cognition in HD patients.
In another ongoing randomized, double-blind controlled clinical study, resveratrol was tested for its preventive activity in 102 early affected HD patients (NCT02336633) [54]. Half of the patients received 80 mg/day of resveratrol, whereas the remaining patients received placebo for 12 months. Clinical benefits of the treatment will be recorded by UHDRS and neuropsychiatric questionnaires. Moreover, the biological tolerance of resveratrol will be evaluated by routine biochemical examination of blood and plasma.
With respect to other antioxidants tested against HD,
Unfortunately, most molecules tested in clinical trials including natural flavonoids failed to provide complete relief or prevent HD, which indicates the disease complexity. In addition, multimodal therapy needs to be tested as it can act on several pathological pathways of HD. As such, therapies including potent antioxidants, pathway blockers, N-methyl-
Although various cellular and animal studies have evaluated the effect of flavonoids on HD models, lack of human trial has limited their therapeutic application. Flavonoids, such as quercetin, naringin, hesperidin, rutin, and isoquercetin, that are likely to prevent and provide symptomatic relief against HD animal models [51,58–61] need to be clinically tested in combination with other promising compounds.
4. THERAPEUTIC STRATEGY FOR HD
The complex mechanisms leading to degeneration and neuronal death in HD are not yet completely elucidated. However, the mutated HTT protein induces anomalies in the DNA and mitochondrial enzymes, with consequent changes in metabolic processes and cellular oxidative stress. Thus, mitochondrial dysfunction and the resulting metabolic and energetic deficits play an important role in the process of HD neurodegeneration [62–64], and these represent an important avenue to explore for effective drug design. The accumulation of the mutated protein within the neurons is responsible for their dysfunction and eventual death [65]; however, it is not yet clear how it disrupts the function of brain cells [66].
Currently, there exists no cure for dementia-related diseases [67], such as HD. One of the most relevant therapeutic strategies is the possibility of delaying the onset of HD when detected early and decreasing the rate of progression. Specific therapeutic approaches are similarly varied and include efforts to reduce the expression of HTT gene, protein accumulation, and protein aggregation [68]. In addition, the possibility of mitigating some symptoms is an important approach in the treatment of HD. Different bioactive molecules are being studied to achieve this goal, one among which are flavonoids.
Although HD is primarily considered a rare neurodegenerative disorder, it has also been linked to glucose metabolism alterations and diabetes, as it has been described in other neurosyndromes such as Friedreich’s ataxia or Alzheimer’s Disease (AD) [69]. Despite having a lower-than-normal body mass index, HD patients tend to have insulin resistance in peripheral tissues and an increased risk for developing Type 2 Diabetes Mellitus (T2DM) [70]. According to an investigation of 610 probands (278 living, 332 deceased) with HD and their first- and second-degree relatives carried out in the National Huntington Disease Research Roster, HD-affected relatives of a diseased proband with diabetes were seven times more likely to have diabetes than non-HD-affected relatives of the proband. Insulin resistance may be a pathogenic factor in HD. One hypothesis for the relationship between insulin resistance and neurodegeneration is that insulin resistance represents a metabolic stressor attending disease progression [71].
Type 2 diabetes mellitus is a chronic endocrine system disease in which the blood levels of glucose in the body cannot be regulated and thus people suffering from this disease have a high level of blood glucose [72]. Dietary flavonoids can prevent and manage T2DM through insulin-dependent approaches, such as protection of pancreatic islet β-cell, reduction of β-cell apoptosis, promotion of β-cell proliferation, attenuation of oxidative stress, activation of insulin signaling, and stimulation of pancreas to secrete insulin, as well as through insulin-independent approaches including inhibition of glucose absorption, inhibition of digestive enzymes, regulation of intestinal microbiota, modification of inflammation response, and inhibition of the formation of advanced glycation end products [73–75] (Figure 5). Flavonoids from coffee, guava tea, whortleberry, olive oil, propolis, chocolate, red wine, grape seed, and cocoa have been reported to show antidiabetic effects in T2D patients through increasing glucose metabolism, improving vascular function, and reducing insulin resistance and HbA1c level [75]. However, individual flavonoid or isoflavonoid appears to have no therapeutic effect on diabetes, although this finding is based on limited clinical data. Preliminary clinical trials have provided evidence that resveratrol exhibits antidiabetic activity in humans by improving glycemic control in patients with insulin resistance [75]. Besides, anthocyanins demonstrated antidiabetic properties by reducing blood glucose and HbA1c levels or by improving insulin secretion and resistance.
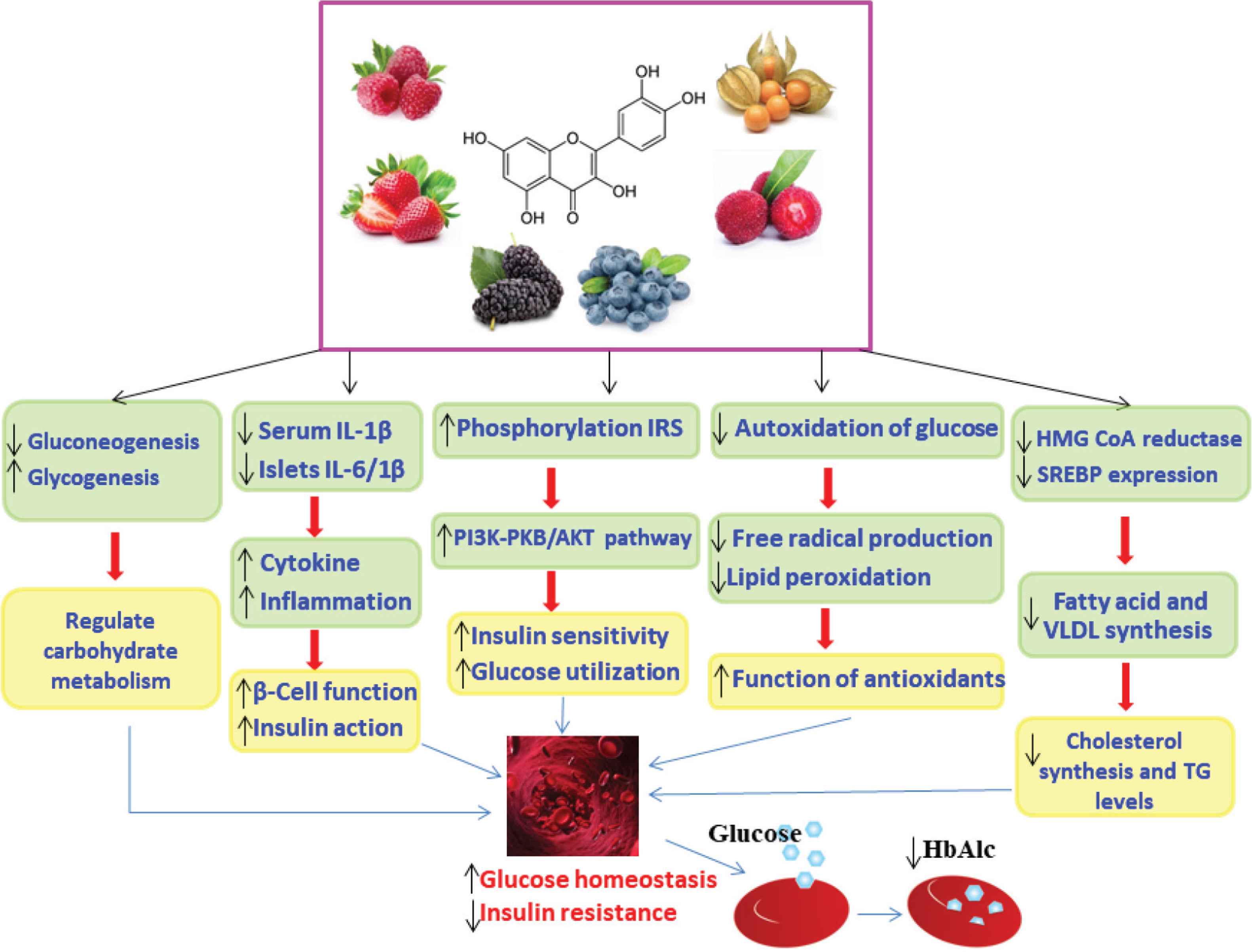
Schematic of the proposed role of flavonoids in the management of blood glucose in diabetes [77]. Flavonoids improve glucose homeostasis by multiple mechanisms including reduction of oxidative stress and inflammatory mediators, decreasing gluconeogenesis, increasing glycogenesis, and altering intracellular signaling pathways. Akt/PKB, protein kinase B; HB, hemoglobin; HbA1c, glycated hemoglobin; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; IL-1β, interleukin-1 beta; IRS, insulin receptor substrate; PI3K, phosphatidylinositol-3-kinase; SREBP-1c, sterol regulatory element-binding protein-1c; TG, triglyceride; VLDL, very-low-density lipoprotein; ↑, increase; ↓, decrease.
Flavonoids prevent the production of free radicals that increase with oxidation of saturated lipids [1]. They also induce vasodilation that involves release of Nitric Oxide (NO) with a subsequent decrease in systolic pressure. Some of them can also promote angiogenesis and all these pathways can be explored as potential treatment options for HD patients. Consuming a flavonoid-rich diet improved vascular health, which is associated with improvement in endothelial function through the activation of vascular endothelial NO synthase and protein kinase B (Akt) [76]. The cardiovascular protection offered by flavonoids in HD is correlated with their antioxidant potential in dementia and the angiogenesis induced that could provide blood nutrients to brain. For instance, tangeretin significantly inhibited the production of proinflammatory cytokines in Lipopolysaccharide (LPS)-stimulated microglia, and this effect was different from other flavonoids that significantly inhibited the production of NO [78]. The evaluation of the antioxidant activity of these kind of molecules need to be more all-inclusive to avoid, or at least to minimize, side effects.
Many flavonoids were, and still are, studied due to their antioxidant potential that helps to regulate oxidants and inflammatory mediators [4]. In addition, the bioactivity potential of flavonoids involves the modulation of cytochrome P450 isoenzymes [77] and the P-Glycoprotein (P-gp)-mediated transport (important efflux transporter). P-gp (now ABCB1) is one of the most extensively studied members of the superfamily of ATP-binding cassette transporters, which are responsible for movement of substances such as ions, sugars, amino acids, phospholipids, peptides, toxins, and antibiotics [79].
In fact, higher P-gp expression was also observed in the brain capillaries of human HD patients. The mutant HTT changed the expression of P-gp through the NF-κB pathway in brain capillaries in HD and markedly affected the availability of P-gp substrates in the brain [80]. Although a wide variety of P-gp inhibitors have been discovered, research efforts are underway to identify the most appropriate one. Flavonoids such as biochanin A, genistein, quercetin, chalcone, silymarin, phloretin, morin, and kaempferol can contribute to P-gp inhibition [81]. The effects produced by some of these components are found to be comparable with those of well-known P-gp inhibitors, such as verapamil and cyclosporine. Thus, flavonoids as P-gp modulators can be included in clinical trials to improve efficacy of drugs as it is already the case for anticancer drugs such as taxanes, anthracyclines, epipodophyllotoxins, camptothecins, and vinca alkaloids [82].
The CYP3A is involved in metabolizing various small molecules from foods and medicinal plants and can be induced or inhibited by some of them too. Until now, few polymorphisms have been reported. However, the frameshift mutation in the human CYP3A subfamily gene CYP3A5, which is created by a T insertion in the nucleotide position 27131-32 and observed in African descendent population, can result in genetic human disorders such as fragile X syndrome or HD that are caused by insertion of CAG that codes for glutamine, creating the so-called Huntington protein, which in turn results in elevated p53 protein level in brain cells that eventually causes cell death by apoptosis. While the number of CAG repeats in the general population may vary from six to 26, patients with HD may have from 40 to over 200, which can grow up to 1000 in some cases [83]. Probably, in the near future, flavonoids could be tested by pharmacogenomic studies to better identify the structures that could be chosen for further research in HD patients. The genetic polymorphism affecting the CYP3A5 enzyme is responsible for interindividual and interethnic variability in the metabolism of CYP3A5 substrates, but this enzyme is expressed only in a limited number of individuals. The absence of CYP3A5 expression in approximately 70% of Caucasians was correlated with a genetic polymorphism (CYP3A5*3) [84].
The influence of the diet rich in flavonoids on mitochondrial metabolism involving in HTT protein should be investigated to a better understanding of the possible treatment. By contrast, it should be noted that although several in vivo and in vitro studies were performed to show the bioactivities of flavonoids, clinical study of flavonoids is quite rare and their mechanism of action has not been fully clarified yet. These compounds might be advantageous because of their less toxic nature in comparison with other synthetic drugs, and wide range of bioactivities, including antiallergic activity, antioxidant activity, antibacterial activity, antidiabetic activity, anti-inflammatory activity, antiviral activity, etc.; however, further investigations are required to determine their exact effects and mechanisms of action.
5. NEUROLOGICAL EFFECTS OF FLAVONOIDS
The pathogenesis of neurodegenerative diseases involves multiple factors. Abnormal protein dynamics, oxidative stress, mitochondrial dysfunction, and environmental factors (such as exposure to pesticides and metal toxicity) have been reported to cause neurodegeneration. Only palliative therapies are available, but none of them is meaningfully able to slow down or halt these diseases (Table 1).
| Drug | Mechanism of action |
|---|---|
| Apomorphine | Dopamine agonism |
| Caprylidene | Increase of the concentration of ketone bodies |
| Dimethyl fumarate | Activation of the NF-E2-related factor 2 pathway |
| Donepezil | Acetylcholinesterase (AChE) inhibition |
| Fingolimod | Blocking the outlet of lymphocytes from lymph nodes |
| Galantamine | AChE inhibition |
| Huperzine A | AChE inhibition |
| Interferon | Modulation of the immune response |
| Memantine | N-methyl- |
| NAMZARIC | NMDA receptor antagonism/AChE inhibition |
| Mitoxantrone | Immunosuppressive action |
| Riluzole | Decrease of glutamate release |
| Rivastigmine | AChE inhibition |
| Ropinirole | Dopamine agonism |
| Rotigotine | Dopamine agonism |
| Teriflunomide | Inhibition of the de novo synthesis of pyrimidine nucleotides, inhibition of T-lymphocyte activation, and cytokine production |
| Tetrabenazine | Disable involuntary writhing movements |
| Tolcapone | Dopamine agonism and COMT inhibition |
COMT, catechol-O-methyltranferase.
Available drugs for the treatment of neurodegenerative disorders
Flavonoids have been showing positive and promising results for the prevention/treatment of neurodegenerative diseases [85,86]. Flavonoids have been demonstrated to modulate neuroinflammation; exert free radicals scavenging activity; chelate transition metals; activate signaling proteins, neuronal receptors, and gene expression survival genes; and regulate mitochondrial functions [87–90]. Specifically, flavonoids modulate the transcription factors NF-κB [91], Phosphatidylinositol 3-Kinase (PI3K)/Akt signaling cascade, and Mitogen-activated Protein Kinase (MAPK) and promote the expression of brain-derived neurotrophic factor. Flavonoids are able to increase the production of NO by inhibiting Cyclooxygenase-2 (COX-2), Inducible Nitric Oxide Synthase (iNOS), and C-reactive protein.
One of the most investigated mechanisms of action of flavonoids is their ability to scavenge ROS [92]. Among them, rutin, genistein, and EGCG weakened 6-hydroxydopamine-induced neurotoxicity in PD [93,94]. Acacetin, myricitrin, and quercetin protected against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity [95–98]. Naringin, hesperidin, and quercetin attenuated 3-Nitropropionic acid (3-NP)-induced neurotoxicity in HD [51,60,61]. Flavonoids are found to interact with critical neuronal signaling pathways including protein kinase C, PI3K/Akt, c-Jun N-terminal Kinase (c-JNK), Extracellular Signal-regulated Protein Kinase 1/2 (ERK1/2), and p38 (cytokinin-specific binding protein) to exert neuroprotective effects [52,99,100]. Dietary flavonoids inhibited LPS-induced production of proinflammatory mediators such as Interleukin-1α (IL-1α), Tumor Necrosis Factor-α (TNF-α), and NO in activated microglia. Catechin, EGCG, quercetin, and kaempferol demonstrated reduced microglia- or astrocyte-mediated neuro-inflammation inhibiting inducible iNOS expression, COX-2, NO production, cytokine release, NADPH oxidase activation, and subsequent ROS generation in activated microglia [101–103].
The prevention of transcription factors activation was also demonstrated. Naringenin reduced the LPS-induced activation of microglial cell through the inhibition of the STAT1 and p38, and the reduction of the expression of iNOS [104,105]. The downregulation of iNOS gene transcription was previously described for quercetin [101].
There is a growing interest in the potential of flavonoids to improve learning, memory, and cognitive function because of their possible effects on molecular and cellular structures [106]. (−)-Epicatechin has been described to induce both cyclic AMP response element-binding protein and ERK1/2 activation in cortical neurons and to enhance the retention of rat spatial memory [91,107].
The anti-inflammatory activity of flavonoids seems to be mediated by their action on artery NO bioavailability, through their potential to inhibit NADPH oxidase and/or to activate endothelial nitric oxide synthase [108–112]. The extent to which these beneficial effects in peripheral arteries might be translatable to beneficial effects in cerebral arteries and improved brain vascularization is presently being studied. Some promising clinical data revealed an increased Cerebral Blood Flow (CBF) that strictly corresponds to the timing of peripheral responses after flavonoids ingestion [113–115]. As such, it has been assumed that the improved human cognitive function observed after flavonoids intake might be partially due to the increased CBF [116]. Indeed, some studies indicated that accentuated declines in brain blood perfusion occur with aging and neurological conditions [117,118].
In summary, the neuroprotective effects of flavonoids involve their antioxidant activity and anti-inflammatory activity; their potential to improve cognitive function, memory, and learning; and their protection of neurons against neurotoxins-induced injury. However, a critical analysis of the literature data revealed that there are only few clinical trials that reported positive results using plant extracts and/or natural compounds to treat neurodegenerative diseases and that there are still no tangible data that can conclusively prove the ability of dietary flavonoids to significantly cross the blood–brain barrier, to access to the brain, and to interact with microglia. Moreover, a limiting factor for the use of flavonoids to treat neurodegenerative diseases is their bioavailability. In this regard, new technologies, including the use of vector-mediated transporters (nanocarriers), are aimed at enhancing the bioavailability and ability of flavonoids to access the brain structures. Future human studies on flavonoids and their metabolites are needed to concretely consider their application in neurodegenerative diseases prevention and/or treatment.
6. FLAVONOIDS AND HD
Various flavonoid derivatives have been evaluated for their therapeutic potentials in HD (Table 2). Some of these compounds are discussed in detail in the following sections.
| Flavonoid | Model | Mechanism/effects | Reference |
|---|---|---|---|
| Hesperidin | 3-NP-induced HD-like symptoms in rats | Hesperidin and naringin diminished the loss in body weight, improved locomotor activity and muscle grip strength, decreased lipid peroxidation and nitrite concentration, and restored superoxide dismutase and catalase activities as compared with the 3-NP-treated group. | [58] |
| Naringin | |||
| Naringin | 3-NP-induced neuronal apoptosis in male Wistar rats | Naringin was found to reverse the 3-NP-induced reduction in enzymic antioxidants activity and level of non-enzymatic antioxidants, and decrease the activity of ATPases in the striatum. Naringin was also observed to reverse the 3-NP-induced reduction in Bcl-2 messenger RNA expression. | [121] |
| Hesperidin | Quinolinic acid-induced HD-like symptoms in male Wistar rats | Whereas hesperidin alone was found to be nonsignificant, the combined administration of hesperidin and minocycline attenuated QA-induced body reduction, impaired behavior, oxidative damage, mitochondrial dysfunction, striatal lesions, and altered levels of expressions of tumor necrosis factor-α, Caspase 3, and brain-derived neurotrophic factor. | [122] |
| Fisetin | PD12 cells expressing mutant Httex1 under the control of inducible promoter | Fisetin activates ERK and reduces the impact of mutant HTT. | [49] |
| Drosophila expressing mutant Httex1 | |||
| R6/2 mouse model of HD | |||
| Quercetin | 3-NP-induced female Wistar rat model of HD | Quercetin reversed 3-NP-induced inhibition of respiratory chain complexes, restored ATP levels, attenuated mitochondrial oxidative stress in terms of lipid peroxidation, and prevented mitochondrial swelling. | [51] |
| Quercetin | 3-NP-induced male Sprague Dawley rat model of HD | Quercetin attenuated 3-NP-induced anxiety, motor coordination deficits, and gait despair. | [123] |
| Quercetin significantly reduced 3-NP-mediated increase in serotonin metabolism, decreased microglial proliferation, and increased astrocyte numbers in the lesion core. | |||
| Rutin | 3-NP-induced rat models of HD | Rutin treatment significantly restored all the biochemical, behavioral, and histological alterations including reduced body weight, locomotor activities, memory, and antioxidants as well as increased levels of lipid peroxides, nitrite, glial fibrillary acidic protein, and activity of acetylcholine esterase. | [124] |
| Myricetin | Binding to mutant RNA | Myricetin was found to interact with the CAG motif of RNA and prevent the translation and sequestration of muscleblind like-1 splicing factor. | [119] |
| Animal models of HD | Myricetin improved neurobehavioral deficits in a mouse model of HD. | ||
| EGCG | In vitro methods | EGCG inhibited the aggregation of mutant HTT exon 1 protein in vitro. | [48] |
| Yeast model of HD | EGCG reduced polyglutamine (polyQ)-mediated HTT protein aggregation in a yeast model of HD. |
3-NP, 3-nitropropionic acid; EGCG, epigallocatechin gallate; HD, Huntington’s disease; QA, quinolinic acid.
Effects of flavonoids in HD models
Hesperidin and naringin are the common flavanone derivatives distributed widely in Citrus plants [119]. Hesperidin and naringin demonstrated protective effects against 3-NP-induced HD-like symptoms in male Wistar rats [58]. Hesperidin and naringin significantly improved locomotor activity and muscle grip strength, and decreased lipid peroxidation and nitrite concentration, and restored Superoxide Dismutase (SOD) and catalase activities as compared with the 3-NP-treated group. Naringin treatment can reverse the 3-NP-induced reduction in enzymic antioxidants activity and level of non-enzymatic antioxidants and decrease in the activity of ATPases in the striatum [120]. Hesperidin in combination with minocycline (a microglial inhibitor) attenuated Quinolinic Acid (QA)-induced HD-like symptoms in male Wistar rats [121].
The supplementation of quercetin was able to reverse 3-NP-induced inhibition of respiratory chain complexes, restore ATP levels, attenuate mitochondrial oxidative stress in terms of lipid peroxidation, and prevent mitochondrial swelling induced by 3-NP in a female Wistar rat model of HD [51]. Quercetin administration along with thiol content also restored the activities of SOD and catalase in 3-NP-treated animals. Quercetin significantly attenuated 3-NP-induced anxiety, motor coordination deficits, and gait despair. Moreover, it provided a significant reduction of 3-NP-mediated increase in serotonin metabolism, decreased microglial proliferation, and increased astrocyte numbers in the lesion core [122]. Rutin has been studied for its potential application as a therapeutic agent for HD. Rutin treatment (25 and 50 mg/kg) significantly restored all the biochemical, behavioral, and histological alterations including reduced body weight, locomotor activities (Rota rod, Open field test), memory (Morris water maze), and antioxidants such as glutathione levels, activities of SOD, catalase, glutathione peroxidase, glutathione-S-transferase, glutathione reductase as well as increased levels of lipid peroxides, nitrite, glial fibrillary acidic protein, and activity of acetylcholine esterase [123]. Fisetin activated ERK in PD12 cells expressing the mutant Httex1 under the control of inducible promoter, Drosophila expressing the mutant Httex1, and R6/2 mouse model of HD [49]. Myricetin was found to interact with the CAG motif of RNA and prevent the translation and sequestration of Muscleblind like-1 (MBNL1) splicing factor [124]. Myricetin also improved neurobehavioral deficits in a mouse model of HD. Ehrnhoefer et al. [48] studied the preventive activity of EGCG on Huntington protein misfolding. EGCG significantly inhibited the aggregation of the mutant HTT exon1 protein in vitro. EGCG also reduced polyQ-mediated HTT protein aggregation in a yeast model of HD.
7. MECHANISM OF FLAVONOIDS FOR HD
The neuroprotective effects of flavonoids are attributed to their antioxidant and anti-inflammatory potential as well as to their ability to modulate various cell signaling pathways. Considering the pathological mechanisms of HD, that is, progressive degeneration of the cerebral cortex and striatal neurons, flavonoids such as quercetin, hesperidin, EGCG, and naringin in various concentrations are proven to be effective agents in both prevention and treatment of HD. It has been suggested that flavonoids can be successfully used for arresting disease progression of HD [52]. Mechanistic targets of flavonoids in the prevention and treatment of HD are summarized in Figure 6.
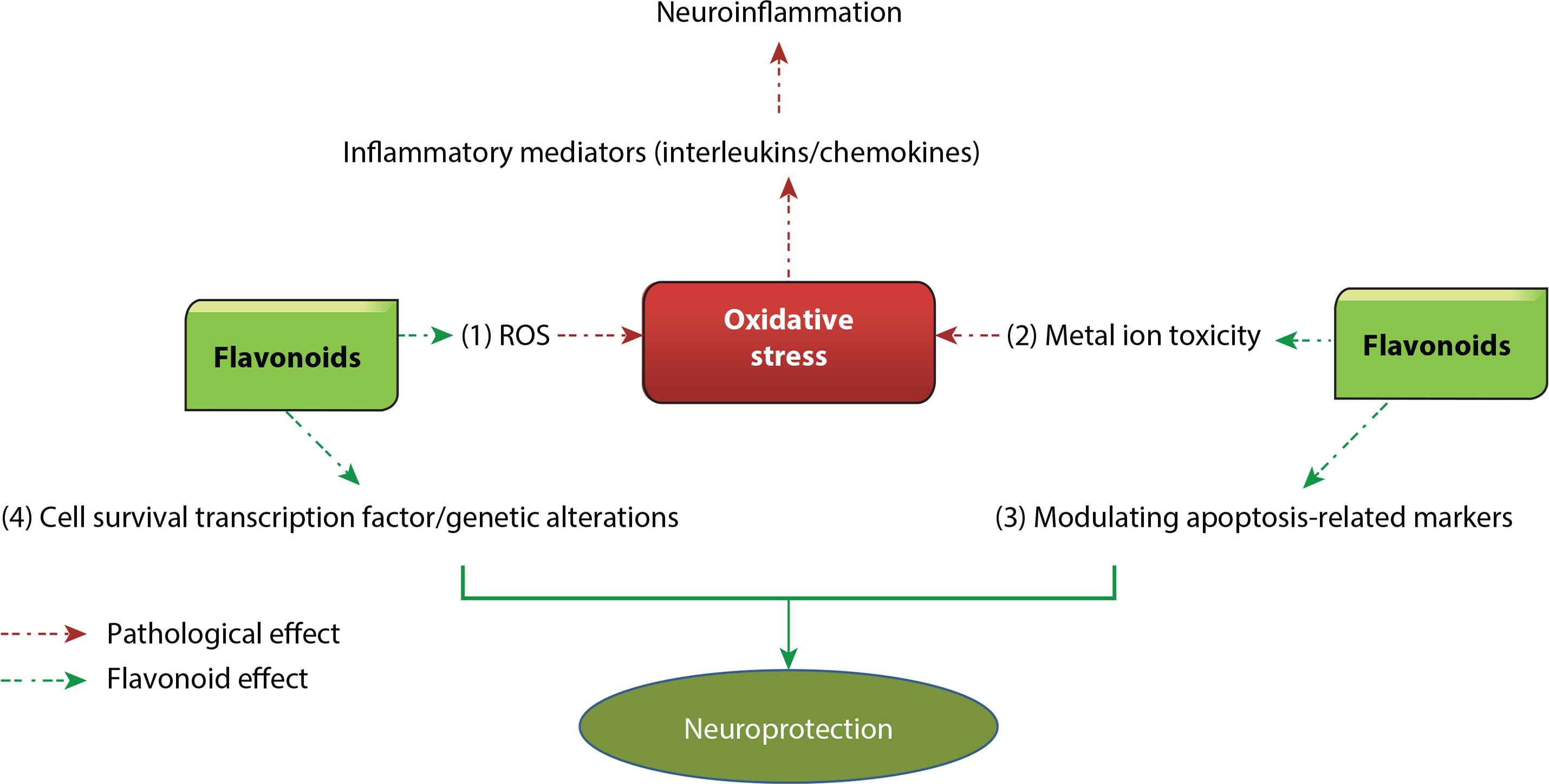
Mechanism of action of flavonoids in Huntington’s Disease (HD). Flavonoids have the potential to treat or prevent HD by targeting multiple mechanisms. (1) Flavonoids can decrease the production of Reactive Oxygen Species (ROS) and increase the production of antioxidant enzymes such as glutathione which leads to reduction of oxidative stress. (2) Metal ion toxicity also enhances oxidative stress in neuronal tissues and flavonoids can chelate metal ions, thus reducing metal ion toxicity. Both these mechanisms help in reduction of oxidative stress which leads to downregulating inflammatory mediators, and thus decrease neuroinflammation, which results in neuroprotection. (3) Flavonoids can modulate apoptosis-related markers including Caspase-3, Bax-Bcl-2, and peroxisome proliferator-activated receptor-γ messenger RNA and (4) can alter genes responsible for causing HD, including those responsible for maintaining mitochondrial activity. They can interact with CAG motifs and alter translation of the huntingtin protein. Both these mechanisms directly result in neuroprotection. Bax, BCL2-associated X protein; Bcl-2, B-cell lymphoma 2.
7.1. Antioxidant and Anti-inflammatory Potential
Flavonoids are the most abundant naturally occurring phenolic compounds well known for their antioxidant properties (Figure 7), which helps in the prevention of a number of diseases including neurodegenerative disorders [125]. The neuroprotective effects of flavonoids are linked to their ability to counter oxidative stress and inflammatory cascade, and their anti-proliferative properties [126]. Naringin showed neuroprotective effects against QA-induced neurotoxicity by modulation of striatal oxidative stress (glutathione, SOD, NO, and malondialdehyde), neuro-inflammatory markers (ILs, TNF-α, and NF-κB), apoptosis-related markers [Caspase-3, BCL2-associated X protein (Bax)-B-Cell Lymphoma 2 (Bcl-2)], and peroxisome proliferator-activated receptor-γ messenger RNA), and mitochondrial complex activity [127]. Khan et al. [128] reported that quercetin possesses antioxidant and anti-inflammatory activities and thus can protect against a range of neurodegenerative disorders. Enogieru et al. [129] reviewed the antioxidant potential of rutin in the protection of neurodegenerative disorders, and documented that the neuroprotective potential of rutin can be attributed to reduction of oxidative stress, inflammatory mediators, restoration of mitochondrial complex enzymes, and upregulation of antiapoptotic genes.
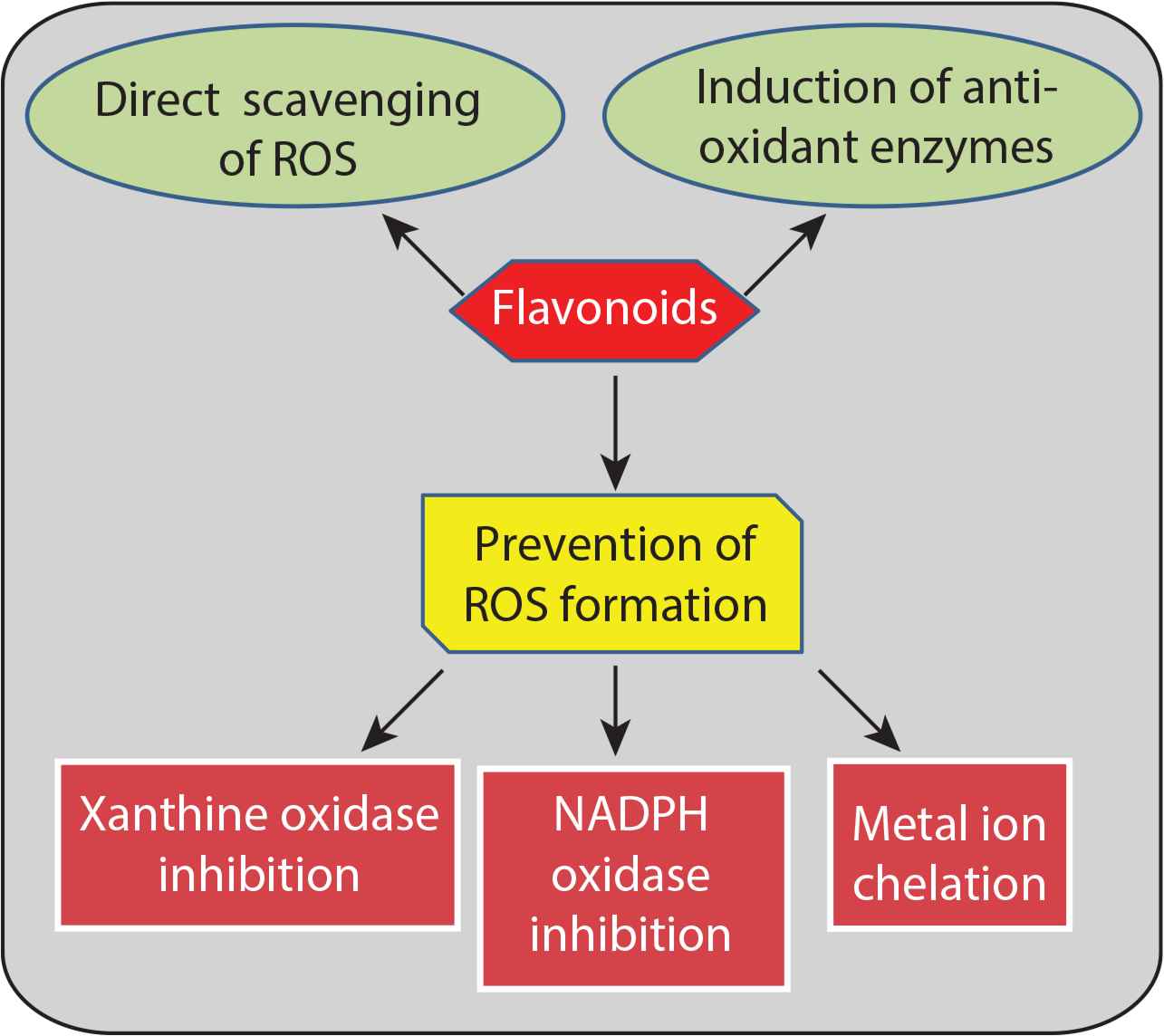
Mechanism of action of antioxidant effects of flavonoids. Flavonoids exert antioxidant effects by Reactive Oxygen Species (ROS) scavenging, preventing ROS formation, and increasing production of antioxidant enzymes.
7.2. Alteration of Intracellular Signaling Pathways
Studies have suggested that flavonoids interact with intracellular signaling pathways, such as p53 and NF-κB, to exert antioxidant, anticancer, and anti-inflammatory activities in neuronal cells which aid in survival of cells in the neuronal circuit (Figure 8) [96,130]. MAPK cascade inhibition by quercetin, rutin, apigenin, and luteolin decreases NO production in activated glial cells, decreasing oxidative stress in neuronal cells and altering mitochondrial apoptotic pathway [131–133]. Activation of the PI3K/Akt pathway by resveratrol to downregulate the inflammatory cascade was reported to be responsible for its neuroprotective effects [134]. Inhibition of Caspase-3, glycogen synthase kinase-3, and JNK and c-jun activation by epicatechin reduce inflammatory mediators and ultimately result in neuroprotection [135,136]. Naringin induces HO-1 expression and enhances nuclear translocation of NF-E2-related factor 2 through the PI3K/Akt signaling pathway, which suggests that naringin augments antioxidant defense capacity and results in the reduction of neurotoxicity [137].
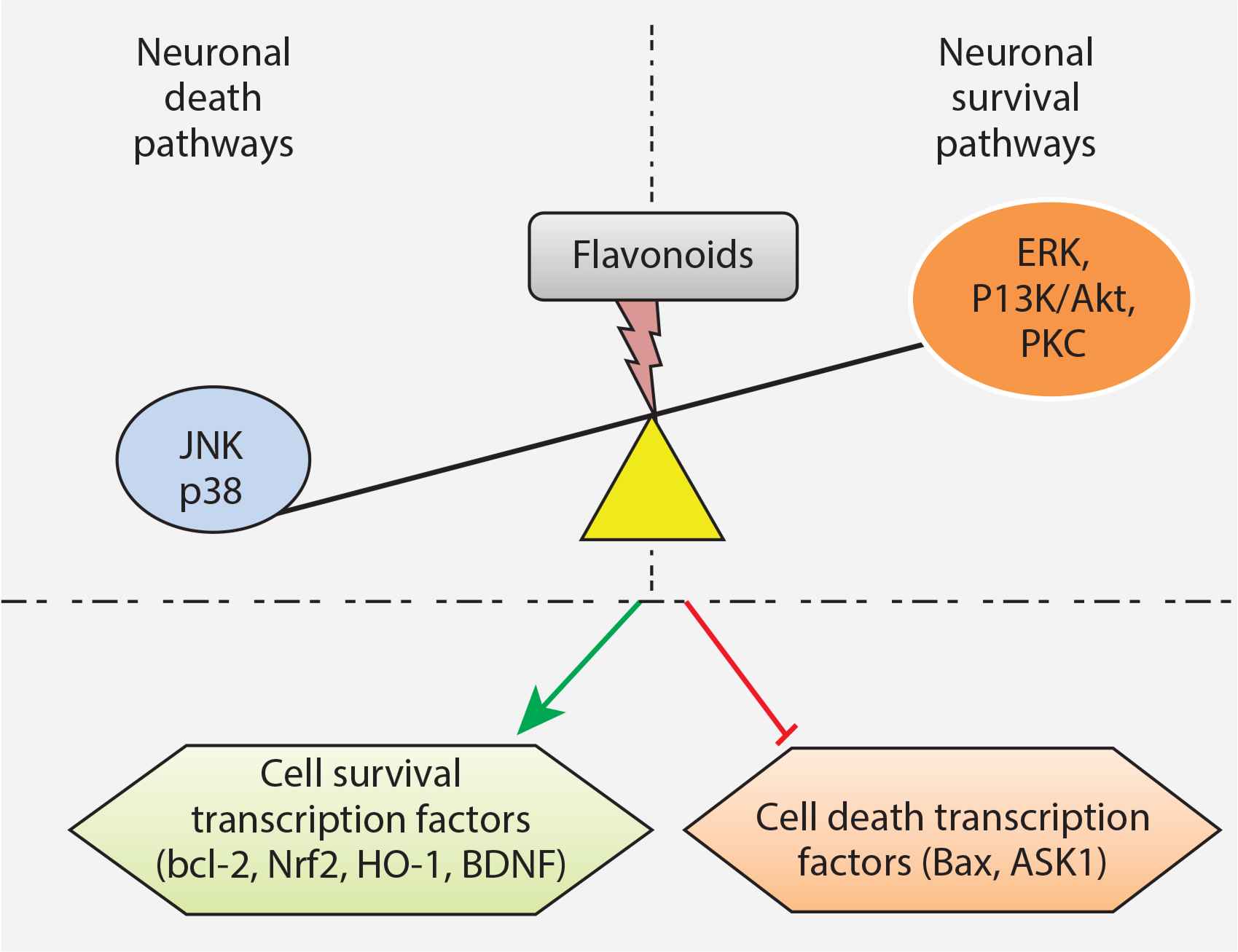
Neuroprotection by flavonoids through modulation of intracellular signaling pathways. Flavonoids activate the neuronal survival pathways and inhibit neuronal death pathways which result in upregulation of cell survival transcription factors and downregulation of cell death transcription factors. ERK, extracellular signal-regulated protein kinase; JNK, c-Jun N-terminal kinase; P13K/Akt, phosphatidylinositol-3 kinase/Akt; PKC, protein kinase C.
7.3. Genetic Alterations
Flavonoids are known to modulate expression of genes, particularly those involved in cell death and survival, which may change synaptic plasticity and alter oxidative as well as inflammatory responses, eventually resulting in protection against degeneration of neurons [45,130,138]. In an HD cell model, resveratrol treatment restored the parameters associated with deregulation of mitochondrial membrane potential and decline in mitochondrial function in a mitochondrial DNA-dependent manner [139]. In YAC128 mice, resveratrol enhanced the expression of mitochondria-encoded electron transport chain genes. Studies indicate that activation of deacetylase improved the transcription of mitochondrial function-associated genes to partially control HD-related motor disturbances. The related literature suggests that pharmacological targeting of histone deacetylases may be of potential benefit in treating neurodegenerative disorders including HD [140]. Myricetin interacting with RNA containing the 5′-CAG/3′-GAC motif via base stacking at AA mismatch prevents the translation of HTT protein and sequestration of MBNL1 [124]. In short, myricetin can be a potential candidate for the prevention of HD through altering translation of HTT protein.
7.4. Metal Ion Chelation
Accumulation of metals such as iron (Fe) and copper (Cu) in the brain can generate oxidative stress which may be a cause of neurodegeneration and can be a pathological hallmark of neurodegenerative disorders such as AD, PD, multiple sclerosis, and HD [141,142]. Polyphenols are reported to protect against Fe- and Cu-induced neurotoxicity via metal chelation [45]. EGCG has greater chelation potential for iron as compared with desferrioxamine along with increasing transferrin receptor protein in SH-SY5Y neuroblastoma cells [143]. Curcumin has been observed to protect against neurodegeneration through its ability to chelate iron [144]. Electron paramagnetic resonance studies reveal that EGCG and gallic acid are ligands for Cu coordination sphere, demonstrating Cu modulatory potential of both these flavonoids [145].
8. CONCLUSION AND FUTURE DIRECTIONS
Current available therapies for the treatment of neurodegenerative disorders including AD, PD, HD, and ALS could not offer complete cure of diseases and are associated with a range of undesirable side effects. Flavonoids are natural polyphenols widely distributed in the plant kingdom and are present in most consumable dietary sources. Having a favorable side effects profile, they can offer an alternative therapeutic choice to conventional therapies in the prevention and treatment of neurodegenerative disorders including HD. In vitro and in vivo studies support the neuroprotective effects of flavonoids (hesperidin, naringin, quercetin, rutin, fisetin, myricetin, luteolin, and EGCG) in different models of neurodegeneration in HD. Flavonoids can modulate multiple mechanisms in neuroprotection by decreasing oxidative stress and inflammatory mediators, altering intracellular pathways, modulating gene expression, and chelating metal ions that are involved in neurodegeneration such as Fe and Cu. Thus, they can be a promising therapeutic choice for the treatment and prevention of HD; however, data from human studies are lacking and clinical trials should be highly encouraged in this regard.
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
AUTHORS’ CONTRIBUTION
HK, HU, RT, TB, HPD, MD, IS, ZC, MGC, EC and MD contributed in writing-original draft preparation, PD and JX contributed in reviewing and editing.
Footnotes
REFERENCES
Cite this article
TY - JOUR AU - Haroon Khan AU - Hammad Ullah AU - Rosa Tundis AU - Tarun Belwal AU - Hari Prasad Devkota AU - Maria Daglia AU - Zafer Cetin AU - Eyup Ilker Saygili AU - Maria da Graça Campos AU - Esra Capanoglu AU - Ming Du AU - Parsa Dar AU - Jianbo Xiao PY - 2020 DA - 2020/02/13 TI - Dietary Flavonoids in the Management of Huntington’s Disease: Mechanism and Clinical Perspective JO - eFood SP - 38 EP - 52 VL - 1 IS - 1 SN - 2666-3066 UR - https://doi.org/10.2991/efood.k.200203.001 DO - 10.2991/efood.k.200203.001 ID - Khan2020 ER -