
Eukaryotic Expression and Purification Technology of Glucosidase in Pichia Pastoris
- DOI
- 10.2991/efood.k.201227.001How to use a DOI?
- Keywords
- Pichia pastoris; glucosidase; eukaryotic expression; protein purification; p-NPG
- Abstract
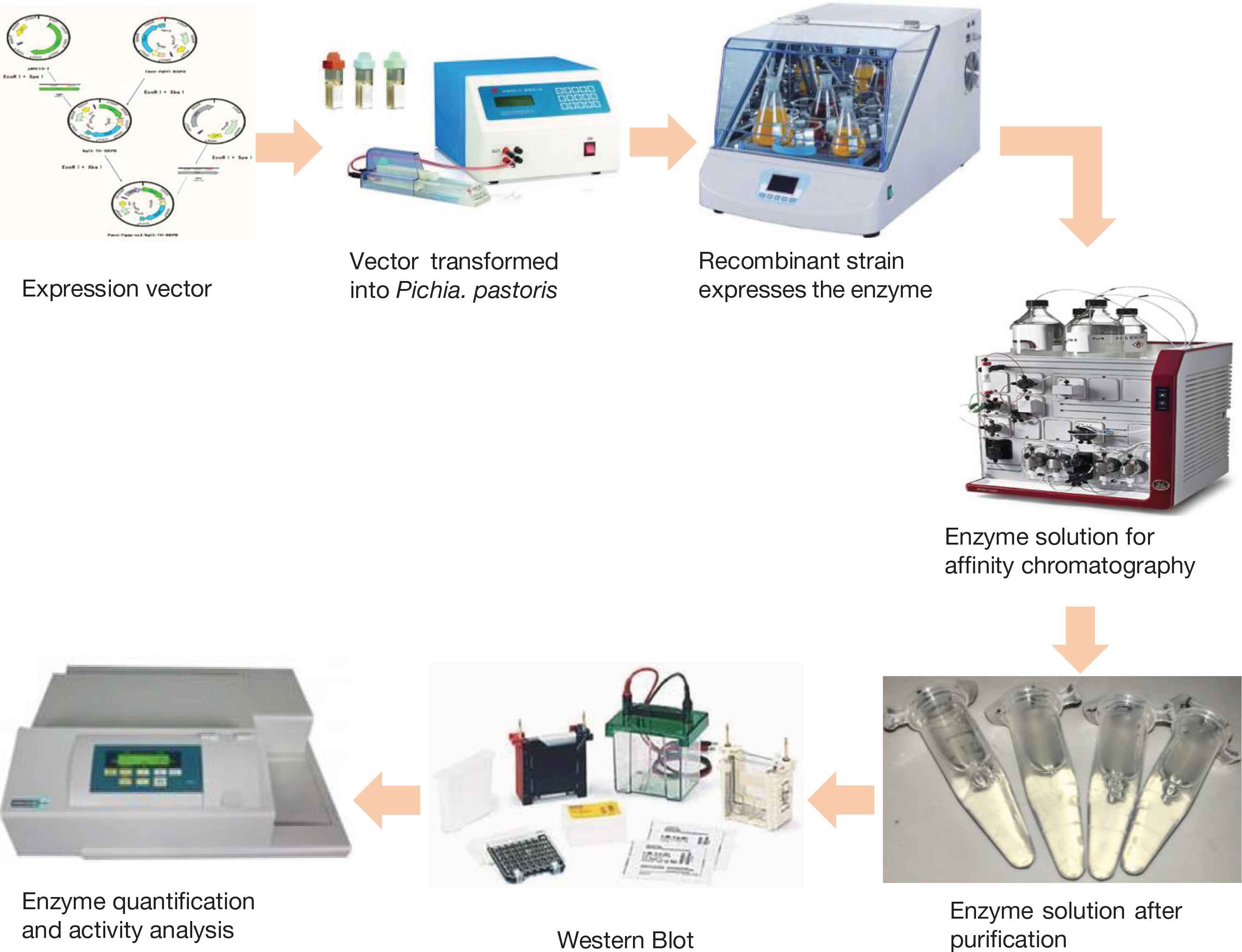
Glucosidase is one of the most important hydrolases in the carbohydrate metabolism pathway in human body. It is noteworthy that glucosidase was raising high attention in numerous researches, attributed to its functional values and biological activities. The expression, secretion, extraction, purification, and quantitative analysis of glucosidase occupies an important position in the research of glucosidase. The Pichia pastoris expression system has been developed for 50 years. With its unique advantages, the expression system has successfully expressed hundreds of heterologous proteins. This article briefly describes the process of high-efficiency expression, extraction and purification of glucosidase in the P. pastoris expression system, focusing on the expression, separation and purification of glucosidase in P. pastoris, as well as the quantitative and activity analysis methods of glucosidase. Affinity chromatography is used for the separation and purification of crude enzyme solution, and UV–visible spectroscopy can be used for enzyme quantification and activity analysis.
- Graphical Abstract
- Copyright
- © 2021 The Authors. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
The main function of glucosidase is to hydrolyze the glycosidic bond and release glucose. Glycosidic bonds are divided into α-type and β-type, so glucosidase also has α-glucosidase and β-glucosidase. In organisms, α-glucosidase is directly involved in the metabolic pathways of starch and glycogen, while β-glucosidase is involved in cellulose metabolism and other physiological pathways. Generally, α-glucosidase is mainly used for the industrial production of Isomalt-Oligosaccharides (IMOs). IMOs are a type of oligosaccharides with a glucose number of 2–5 and at least one α-1,6 glycosidic bond. IMOs have the effects of preventing dental caries, lowering blood pressure, and lowering cholesterol. Therefore, glucosidase is widely applied in the food and pharmaceutical industries [1].
At present, there are many sources of glucosidase, including plants, mammals, fungi, and bacteria. However, the yield of glucosidase produced by natural strains is low and it is difficult to extract and purify. For example, Aspergillus niger is an excellent strain for producing α-glucosidase, but its low yield and complicated purification steps are the biggest disadvantages.
The Pichia pastoris yeast expression system was first publicly released in 1969 [2]. It has been achieved tremendous development in the past 50 years. Hundreds of heterologous proteins have been successfully expressed in P. pastoris [3]. The simple and practical high-density culture method overcomes the shortcomings that prokaryotic expression systems such as Escherichia coli expression systems are prone to produce insoluble inclusion bodies, and it also overcomes other eukaryotic expression systems such as mammalian cell expression systems that require strict operating environments. At the same time, P. pastoris has high biological safety and is suitable for the production of many food enzymes and protein drugs [4].
In the research of glucosidase, the expression, secretion, extraction, purification, and quantitative analysis of glucosidase are essential. This article briefly summarizes the methods of glucosidase expression, isolation and purification, and detection of enzyme activity in P. pastoris in order to provide a useful summary for the research community.
2. PICHIA PASTORIS EXPRESSION SYSTEM
The advantages of P. pastoris expression system were listed as followed: (1) Use alcohol oxidase as a powerful promoter to control the expression of foreign proteins; (2) The heterologous gene is directly integrated into the yeast chromosome, so the engineered strain is very stable; (3) Yeast performs post-translational modification of heterologous protein to give the protein its original activity; (4) Low fermentation cost and easy large-scale fermentation; (5) P. pastoris itself secretes fewer proteins, so it is easy to separate and purify the heterologous expression target protein [4].
3. CODON OPTIMIZATION STRATEGY
In order to ensure the smooth expression of heterologous genes in P. pastoris, codon optimization of heterologous genes occupies a crucial position [5]. Due to the codon preference and degeneracy of codons in different organisms or different proteins of the same species, heterologous genes are used differently in the host with the same codon. If the heterologous gene is directly expressed in the host cell, it is possible that the secondary structure stability of the mRNA will decrease due to the codon preference, which affects the entire translation process from the initiation and extension levels of translation. For the host, the more rare codons a heterologous gene contains, the more difficult it is to express the heterologous protein [6]. The current methods of codon optimization include optimizing host cells and optimizing target genes.
4. SELECTION OF HETEROLOGOUS GENE EXPRESSION VECTOR COMPONENTS
Most foreign genes will have their own signal peptide sequence, but this is generally not suitable for expression in heterologous hosts, and a suitable signal peptide (such as α signal peptide) can be selected instead. Second, the copy number of the heterologous gene can be increased to increase the expression level of the heterologous gene in the host cell [7]. Finally, add a suitable tag such as His tag to facilitate the separation and purification of foreign proteins.
5. ACQUISITION OF TARGET GENE AND CONSTRUCTION OF EXPRESSION VECTOR
According to the gene sequence of glucosidase in the NCBI database, add Pgap promoter and α-signal peptide to the N-terminal of the gene coding sequence, add 6×His tag to the C-terminal and finally add Sac I and EcoR I restriction sites at both ends of the sequence. Use codon usage preferences to optimize the sequence and perform chemical synthesis, insert it into the pPIC3.5K expression vector to obtain an expression vector containing foreign genes.
6. CONSTRUCTION AND SCREENING OF RECOMBINANT BACTERIA
There are generally three methods to transform the recombinant plasmid containing the expression vector of the foreign gene into Pichia competent cells: P. pastoris lithium chloride transformation method, PEG1000 transformation method and P. pastoris electroporation method [8].
The host strain P. pastoris (GS115/SMD1168/KM71) is a histidine-deficient (his-) strain and cannot grow on Minimal Dextrose medium (MD medium), when the expression vector plasmid containing the foreign gene is integrated into the P. pastoris chromosome, the his4 gene on the expression vector is complementary to the his4 gene of the P. pastoris genome to form a complete his4 gene [8].
The recombinant plasmid was linearized with Sal I enzyme and transformed into P. pastoris GS115 competent cells by electroporation. After electroporation, the yeast solution was added to MD medium for culture and selection of positive transformants [9]. Confirm whether the transformant contains the glucosidase gene or not by polymerase chain reaction (PCR) verification.
Electrical transfer instrument parameters: voltage: 1500 V, resistance: 400 Ω, capacitance: 25 μF, pulse time: 5 ms, one electric shock.
7. EXPRESSION OF THE RECOMBINANT IN PICHIA PASTORIS
The recombinant yeast was inoculated into a test tube containing 5 mL YPG liquid medium (1% yeast extract, 2% peptone, 1% glycerol) for activation, cultured at 29°C for 24 h, then inoculated into a 1 L shake flask containing 300 mL YPG medium, and cultured at 29°C for 72 h. The culture supernatant of transformed P. pastoris is glucosidase.
8. PURIFICATION OF GLUCOSIDASE
Because the His tag is added to the glucosidase protein, the protein can firmly bind to Ni2+ on the agarose affinity column. In addition, imidazole can competitively bind to Ni2+, and the imidazole solution can elute glucosidase from the column. To achieve the effect of separation and purification [10].
8.1. Preparation of Mobile Phase
Solution A: 50 mM PBS, 500 mM NaCl, 10 mM imidazole, pH = 7.4, and filtered with 0.22 μm membrane.
Solution B: 50 mM PBS, 500 mM NaCl, 250 mM imidazole, pH = 7.4, and filtered with 0.22 μm membrane.
8.2. Preparation of Loading Solution
Ammonium sulfate powder was slowly added into the culture supernatant until the final concentration of ammonium sulfate reaches 80% (56.1 g ammonium sulfate powder was added into 100 mL supernatant), keep it at 4°C overnight. The precipitate obtained by centrifugation is the ammonium sulfate precipitate. Use solution A to resuspend the precipitate. After centrifugation of the resuspension, take the supernatant and filter with filter paper.
8.3. Affinity Chromatography
The filtrate was injected into the histidine affinity chromatography column (Ni Sepharose TM Fast Flow) that has been pre-equilibrated with solution A in the AKTA pure. After the filtrate was injected, the column is re-equilibrated with solution A, followed by gradient elution with 30–50% solution B, collecting elution peaks, and performing electrophoresis detection and enzyme activity detection.
9. SDS-PAGE AND WESTERN BLOT DETECTION
Sodium dodecyl sulfate-polyacrylamide gel electrophpresis (SDS-PAGE) was performed using a 10% (w/v) polyacrylamide gel with a 5% stacking gel and the Mini-Protean II system (Bio-Rad, California, USA) [11]. Because of the presence of the histidine tag, western blotting was used to detect the glucosidase protein by adding an anti-histidine tag antibody.
10. PROTEIN CONTENT DETERMINATION
The Bradford method was used to determine the protein content in the enzyme solution, and the standard protein solution was used to make a standard curve. Mix 250 μL of Coomassie Brilliant Blue G-250 with 5 L of enzyme solution, stand for 5 min for color development, measure the absorbance at 595 nm with a microplate reader, and calculate the protein concentration according to the standard curve.
11. DETERMINATION OF GLUCOSIDASE ACTIVITY
11.1. Principle
Under the action of glucosidase, p-nitrophenyl-α-
11.2. Standard Curve
According to the absorbance values of different concentrations of pNP solutions at 405 nm, draw a standard curve, with the concentration as the abscissa and the absorbance as the ordinate.
11.3. Measurement Procedure
Take 900 μL of Na2HPO4-citrate buffer (0.2 mol/L, pH = 5) and 50 μL of pNPG (0.1 mol/L) at 50°C, add 50 μL of enzyme solution to be tested, and react at 50°C for 10 min. Add 1 mL of pre-cooled Na2CO3 solution (1 mol/L) to stop the reaction and develop color. Use a microplate reader to measure the absorbance at 405 nm. Calculate the pNP concentration produced according to the standard curve.
11.4. Enzyme Activity Calculation Formula
C: The concentration of pNP substituted into the standard curve (μmol/mL)
V1: reaction volume (mL)
N: dilution factor
V2: volume of enzyme added (mL)
t: reaction time
12. CONCLUSION
Glucosidase has been widely studied as an important industrial enzyme for the industrial production of IMOs. The efficient and simple expression, isolation and purification of this enzyme are the basis of numerous studies. Traditionally, the properties of glucosidase products, activity of glucosidase, and protein concentration were analyzed base on the absorbance of specific wavelength, such as high performance liquid chromatography (HPLC) and HPLC–mass spectrometry (HPLC-MS).
Glucosidase is mostly derived from eukaryotes and is essentially glycoprotein. The E. coli expression system cannot perform post-translational modification and processing. The P. pastoris expression system has post-transcriptional processing and modification functions, which is suitable for stable expression of functional foreign proteins. Compared with insect expression systems and mammalian expression systems, P. pastoris expression systems are simple to operate, low in cost, and can be fermented on a large scale. It is an ideal recombinant eukaryotic protein production and preparation tool.
This article introduces the use of P. pastoris expression system to express and purify to obtain active and high-purity glucosidase, which is the basis for future research on the biochemical properties of glucosidase.
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
AUTHORS’ CONTRIBUTION
WZ contributed in investigation, resources, methodology, writing – review & editing. DY contributed in investigation, supervision. WB contributed in conceptualization, supervision, project administration.
Footnotes
REFERENCES
Cite this article
TY - JOUR AU - Weijie Zhou AU - Dongsheng Yao AU - Weibin Bai PY - 2021 DA - 2021/01/08 TI - Eukaryotic Expression and Purification Technology of Glucosidase in Pichia Pastoris JO - eFood SP - 406 EP - 409 VL - 1 IS - 6 SN - 2666-3066 UR - https://doi.org/10.2991/efood.k.201227.001 DO - 10.2991/efood.k.201227.001 ID - Zhou2021 ER -